
Direct reductive coupling of indoles to nitrostyrenes en route to (indol-3-yl)acetamides†
Michael
Rubin  *ad
*ad
*ad
Abstract
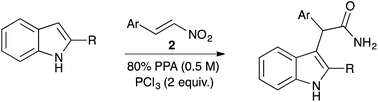
A highly efficient one-pot method for the reductive coupling of indoles to nitrostyrenes in polyphosphoric acid doped with PCl3 was developed. This method allows direct and expeditious access to primary (indol-3-yl)acetamides, interesting as anti-cancer drug candidates.
 Please wait while we load your content...
Please wait while we load your content...

