DOI:
10.1039/C6RA15186H
(Paper)
RSC Adv., 2016,
6, 93867-93880
Ag(I), Cu(II), Co(III) and Hg(II) complexes and metal-assisted products derived from 4-methyl-piperidine-carbodithioate: syntheses, structures, thermal analyses, redox behaviour and fluorescence properties†
Received
11th June 2016
, Accepted 15th September 2016
First published on 15th September 2016
Abstract
Four new complexes [Ag2(4-mpipdtc)2(PPh3)2] (1), [Cu(4-mpipdtc)2] (2), [Co(4-mpipdtc)3]·CHCl3 (3) and [PhHg(4-mpipdtc)] (4) and two new products, bis(4-methyl piperidinethiocarbonyl) disulfide {(4-mpipdtc)2} (5) and (4-methyl-piperidin-1-yl) carbothioylsulfanyl-methyl (4-methyl)-piperidine-1-carbodithioate {CH2(4-mpipdtc)2} (6) have been obtained in this study. The syntheses of compounds 5 and 6 were assisted by Mn(II) and Ag(I) ions, respectively, and were obtained from potassium 4-methyl-piperidine-carbodithioate {K+(4-mpipdtc)−} {where, 4-mpipdtc− = 4-methyl piperidine carbodithioate}. All new compounds have been characterized by elemental analyses, IR, NMR, magnetic susceptibility and single X-ray crystallography techniques. These compounds are stabilized by intermolecular C–H⋯S, S⋯S, C–H⋯π and C–H⋯N interactions. The most interesting feature in complex 4 is that the ligand bound phenylmercury cation is stabilized via intermolecular as well as intramolecular Hg⋯S secondary interactions. Compounds 3 and 5 are highly fluorescent in a solution when compared to the free ligand and emit violet/violet-blue light at 372 and 413 nm upon excitation at 328 and 300 nm, respectively. The course of the thermal degradation of complexes 1–4 have been investigated by TG-DTA, which indicates that metal sulphide is formed as the final residue. The results obtained from the electronic structure calculations at the density functional theory level corroborate our experimental findings obtained from the IR data. Frontier molecular orbital analysis reveals that complexes 1 and 3 are softer and more reactive than complexes 2 and 4. Cyclic voltammetry shows that complex 2 exhibits a reversible Cu(II)/Cu(I) redox process at 0.515 V, whereas the ligand and complexes 1, 3 and 4 show irreversible redox behaviour.
1. Introduction
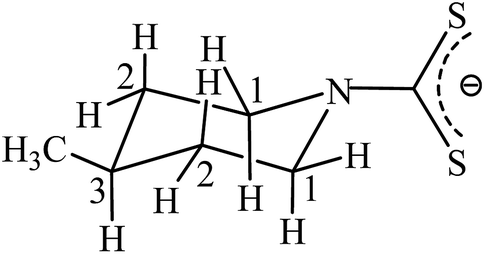
Dithiocarbamates are versatile ligands capable of forming a variety of complexes with most of the transition elements in various oxidation states. Metal complexes of dithiocarbamato ligands have been attracted a lot of attention in the field of coordination and organometallic chemistry due to their applications in molecular electrical conductivity, optical, magnetic properties and as a single source precursor for the preparation of metal sulphide nanoparticles. They are also applicable in industrial areas such as rubber vulcanization accelerators, flotation agents in metallurgy and petroleum additives, and have importance in biological processes.1–10 Ag(I) dithiocarbamato complexes have extensively been used in the formation of Ag2S nanoparticles and nanoscale metals.11 Cobalt(III) dithiocarbamato complexes have been used as catalysts in organic transformations such as the enamination reaction.12 Furthermore, fluorescent nanowires of cobalt(III) dithiocarbamato complexes have been synthesized and used in optoelectronic devices.13 Organomercury(II) dithiocarbamato complexes exhibit a great affinity for sulfur donor sites present in protein frameworks.14–16 Piperidine dithiocarbamato ligands generally form four membered S,S-chelate rings and bridge two metal centres due to the dominant contribution of the resonance form III (Scheme 1).17 The thiol form may undergo aerial oxidation to form a disulphide or coupling of two thiol groups via a metal-catalyzed reaction. The most common route for creating disulphide bonds is via the oxidation of thiol groups, which provides stability to protein dimers, polymers and complexes in which the sulphide bonds helps in protein folding.18 Metal ion-assisted cyclization of thiosemicarbazides into their corresponding oxadiazole/thiadiazole and disulphide bond formation have been reported.19,20 The synthesis of bis(dialkylaminethiocarbonyl) disulfides has been reported but their structural characterization has not been performed. Moreover, these syntheses involve sodium nitrate and hydrochloric acid, which resulted in poor yields of the products.21 Disulfide bond formation of methylene bis-dithiocarbamates has been reported either in the absence21–23 or the presence of metal salts.24 Cyclic and open-chain amines react with carbon disulfide and methylene halides in the presence of an ionic liquid ([pmIm]Br) at room temperature to produce the corresponding dithiocarbamates.23 These symmetrical bis-dithiocarbamates are used further as valuable intermediates in the synthesis of several biologically active molecules.25 Some complexes of the methyl-piperidine carbodithioates that have been reported are studied on the basis of their electronic and infrared spectra but no data pertaining to single X-ray crystallography is available.26 In view of the interesting bonding modes and various properties as discussed above, herein we report on the syntheses, photoluminescence, thermal behaviour and structural characterization of Ag(I), Cu(II), Co(III) and Hg(II) complexes of the 4-methyl piperidine dithiocarbamato ligand. Additionally, the formation of the disulfide bridged product 5 obtained using Mn2+ and another product 6 containing the S–CH2–S group, catalysed by Ag+ ions are also reported.
 |
| | Scheme 1 The resonance structures of 4-methyl-piperidine-carbodithioate. | |
2. Results and discussion
Compounds 1–6 were synthesized in a mixture of methanol–chloroform via a simple and very convenient method at room temperature. These compounds have been characterised by various physiochemical techniques. For the structural studies, crystals of 1–6 were grown at room temperature using a slow solvent evaporation technique. A methanol solution of the ligand, 4-methyl-piperidine-carbodithioate reacted with AgNO3 to form a white precipitate, which dissolved upon the addition of a chloroform solution of PPh3 yielding [Ag2(4-mpipdtc)2(PPh3)2] (1). A methanol-chloroform solution of the ligand reacted with Cu(OAc)2·H2O/CoCl2/PhHgOAc to form clear solutions yielding the complexes [Cu(4-mpipdtc)2] (2), [Co(4-mpipdtc)3]·CHCl3 (3) and [PhHg(4-mpipdtc)] (4), respectively. Scheme 2 depicts the formation of complexes 1–4 in which complex 1 contains triphenylphosphine as a co-ligand. {(4-mpipdtc)2} (5) and {CH2(4-mpipdtc)2} (6) were synthesized via the reaction of the 4-methyl-piperidine-carbodithioate with Mn(OAc)2·4H2O and AgNO3 in methanol and methanol–dichloromethane mixture, respectively, at room temperature with simple stirring of the reagents. Scheme 3 depicts the formation of compounds 5 and 6. We have synthesized bis(dialkylaminethiocarbonyl) disulfide from the potassium salt of dithiocarbamates assisted by Mn(II) acetate at room temperature, this method is different from the reported synthesis.21 Both the syntheses are simple one-pot and multi-component reactions completed in two steps. The present route is relatively safer involving less hazardous chemicals that can be easily handled. Furthermore, the formation of symmetrical methylene bis(dithiocarbamates) reported in the literature have been the by-products of the reaction between transition metal halides and sodium or potassium salts of dithiocarbamates when methylene chloride was used as the solvent. Generally, methylene chloride can react with strong nucleophiles such as the sodium salt of thiols to give the corresponding symmetrical bis-dithio compounds, however, it requires long reaction times and gives low product yields. In the present reaction, methylene chloride acts as an electrophile and the sodium/potassium salt of thiols/dithiocarbamates acts as strong nucleophiles. Although the reaction proceeded equally well with CH2Cl2, CH2Br2 and CH2I2 for the synthesis of compound 6, CH2Cl2 was preferred due to its widespread availability and low cost. Our work reports the silver(I) nitrate assisted synthesis of symmetrical bis-dithiocarbamates in the presence of dichloromethane with product yields in the range of 80–90%. Complex 1 is soluble in chloroform and DCM and complex 2 is soluble in chloroform and DMSO. Compounds 2, 3, 5 and 6 are soluble in methanol, acetonitrile and DMSO. Compounds 1, 2, 3, 4, 5 and 6 melt at 196, 213, 309, 248, 102 and 140 °C, respectively.
 |
| | Scheme 2 Syntheses of complexes 1–4. | |
 |
| | Scheme 3 Syntheses of the Mn(II)- and Ag(I)-assisted products 5 and 6. | |
2.1 Magnetic moments and electronic spectra
Complexes [Ag2(4-mpipdtc)2(PPh3)2] (1), [Co(4-mpipdtc)3]·CHCl3 (3) and [PhHg(4-mpipdtc)] (4) are diamagnetic indicating the presence of low spin Ag(I), Co(III) and Hg(II) centres. Complexes 1, 3 and 4 showed absorptions in the region of 263–328 nm due to intra-ligand/charge transfer transitions Fig. 1(a). The complex, [Cu(4-mpipdtc)2] (2) showed a magnetic moment of 1.75 BM, which indicates the presence of an unpaired electron. The presence of a broad band around 439 nm was assigned to the envelope of the 2B1g → 2A1g, 2B2g and 2Eg transitions and suggests a square planar geometry for the complex (ESI Fig. 1†). Another high energy band observed at 272 nm was assigned to π → π intra-ligand/charge transfer transitions.27
 |
| | Fig. 1 (a) The absorption spectra of the compounds at 2 × 10−5 M and (b) the emission spectra of the compounds at 2 × 10−5 M excitation wavelength was 300 nm for compounds 1–6, and the ligand and also at 328 nm for compound 3 in graph 3(b). | |
2.2 IR spectra
The IR spectrum of the potassium derivative of the 4-methyl-piperidine-carbodithioate ligand, {K+(4-mpipdtc)−}, showed absorptions due to the stretches attributed to C–H (2951 cm−1), C–N (1585 cm−1) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S (1015 cm−1) bonds, respectively. The IR spectra of [Ag2(4-mpipdtc)2(PPh3)2] (1), [Cu(4-mpipdtc)2] (2), [Co(4-mpipdtc)3]·CHCl3 (3), [PhHg(4-mpipdtc)] (4), {(4-mpipdtc)2} (5) and {CH2(4-mpipdtc)2} (6) showed aliphatic ν(C–H) bands at 2989, 2948, 2949, 2947, 2950, 2924 cm−1, respectively. The aromatic ν(C–H) band for 1 and 4 appeared at 3058 and 3062 nm, respectively. The IR spectra of compounds 1–6 revealed ν(C–N) bands at 1469, 1504, 1493, 1484, 1481 and 1434 cm−1, respectively. The ν(CS) band was observed at 962, 965, 966 and 968 cm−1 for complexes 1–4, respectively. A new band for complexes 1–4, attributable to ν(M − S) appeared at 320 (calc.), 429, 428 and 430 cm−1, respectively. The ν(CS) band suffered a negative shift of 53, 50, 49 and 47 cm−1 in complexes 1, 2, 3, and 4, respectively, when compared to the free ligand indicating the involvement of the dithio sulfur atoms in bonding. Compounds 5 and 6 showed two bands at 959, 816 and 998, 714 cm−1, respectively indicating the presence of C–S and CS bonds (Table 1) (ESI Fig. 2–8†).
S (1015 cm−1) bonds, respectively. The IR spectra of [Ag2(4-mpipdtc)2(PPh3)2] (1), [Cu(4-mpipdtc)2] (2), [Co(4-mpipdtc)3]·CHCl3 (3), [PhHg(4-mpipdtc)] (4), {(4-mpipdtc)2} (5) and {CH2(4-mpipdtc)2} (6) showed aliphatic ν(C–H) bands at 2989, 2948, 2949, 2947, 2950, 2924 cm−1, respectively. The aromatic ν(C–H) band for 1 and 4 appeared at 3058 and 3062 nm, respectively. The IR spectra of compounds 1–6 revealed ν(C–N) bands at 1469, 1504, 1493, 1484, 1481 and 1434 cm−1, respectively. The ν(CS) band was observed at 962, 965, 966 and 968 cm−1 for complexes 1–4, respectively. A new band for complexes 1–4, attributable to ν(M − S) appeared at 320 (calc.), 429, 428 and 430 cm−1, respectively. The ν(CS) band suffered a negative shift of 53, 50, 49 and 47 cm−1 in complexes 1, 2, 3, and 4, respectively, when compared to the free ligand indicating the involvement of the dithio sulfur atoms in bonding. Compounds 5 and 6 showed two bands at 959, 816 and 998, 714 cm−1, respectively indicating the presence of C–S and CS bonds (Table 1) (ESI Fig. 2–8†).
Table 1 The important IR bands (cm−1) for the {K+(4-mpipdtc)−} ligand and compounds 1–6
| Compound |
ν(C–H) |
ν(C–N) |
ν(CS/C–S) |
ν(M − S) |
| K+(4-mpipdtc)− |
2951 |
1585 |
1015 |
— |
| [Ag2(4-mpipdtc)2(PPh3)2] (1) |
3058, 2989 |
1469 |
962 |
320 |
| [Cu(4-mpipdtc)2] (2) |
2948 |
1504 |
965 |
429 |
| [Co(4-mpipdtc)3]·CHCl3 (3) |
2949 |
1493 |
966 |
428 |
| [PhHg(4-mpipdtc)] (4) |
3062, 2947 |
1484 |
968 |
430 |
| (4-mpipdtc)2 (5) |
2950 |
1481 |
959/816 |
— |
| CH2(4-mpipdtc)2 (6) |
2924 |
1434 |
998/714 |
— |
2.3 NMR studies
The 1H NMR spectra of {K+(4-mpipdtc)−}, 1, 3, 4, 5 and 6 showed a triplet at δ 3.76, 3.07, 2.89, 3.10, 3.32 and 2.95 ppm, respectively due to the axial proton C1H. The signal for the equatorial protons C1H appeared as a doublet at δ 5.67, 5.30, 4.66, 4.95, 5.00 and 4.09 ppm for {K+(4-mpipdtc)−}, 1, 3, 4, 5 and 6, respectively. For the equatorial protons, only the geminal coupling with the axial protons was large but in the case of the axial protons both the geminal coupling with the equatorial protons as well as the vicinal coupling with the adjacent axial protons are both large/significant. The deshielding of the equatorial and axial protons is due to the high electron density at the nitrogen atom,28,29 however, the equatorial proton (H-1e) is more deshielded than the axial proton (H-1a).30 The signals for the C2H and C3H protons appear as multiplets in the range of δ 1.20–1.75 and δ 1.68–1.79 ppm, respectively. A doublet for the methyl protons appeared in the range δ 0.93–0.99 ppm for all the compounds.31 The singlet for the SCH2 protons is highly deshielded due to the presence of the sulfur atom around the protons and appeared at δ 5.37 ppm in compound 6.24
The 13C NMR spectrum of the {K+(4-mpipdtc)−} ligand showed a signal at δ 208.76 ppm due to the CS2 carbon. Complexes 1, 3 and 4 showed a signal for the NCS2 carbon at δ 207.17, 203.55 and δ 200.81 ppm, respectively. This indicates the considerable double bond character in the nitrogen carbon bond of the dithiocarbamato group. A considerable δ+ surplus charge is localized on the nitrogen atom and a δ− charge is delocalized via four-membered metallochelate ring –CS2M.30–32 The CS2 carbon for compounds 5 and 6 appeared at δ 192.82 and 193.62 ppm, respectively, which are less downfield shifted than the free ligand as well as in complexes 1, 2, 3 and 4. The carbon atom (C1) adjacent to the nitrogen of dithiocarbamate was more deshielded than other carbon atoms in the piperidine ring (Scheme 4) and appeared at δ 51.29, 52.74, 45.48, 52.88, 47.43 and 52.04 ppm for {K+(4-mpipdtc)−}, 1, 2, 3, 4, 5 and 6, respectively. The C3 carbon is more deshielded than the C2 carbon and the signals for C3 and C2 were observed at δ 34.35, 30.89 {K+(4-mpipdtc)−}; 33.87, 30.39 (1); 33.13, 30.65 (3); 33.79, 30.25(4); 33.83, 30.85 (5) and 34.00, 30.83 (6) ppm.29 The methyl carbon was observed in the range δ 21.19–21.56 ppm for all the compounds. In compound 6, a new signal observed at δ 54.52 ppm was due to the SCH2S carbon.24 The CS signal was observed in the range of 200.81–207.17 ppm and were found to be shielded when compared to the free ligand (δ 208.76 ppm) due to the involvement of CS2 in bonding.
 |
| | Scheme 4 Numbering of the ligand moiety. | |
2.4 Photoluminescence studies
The emission spectra of compounds 1−6 were recorded in methanol and chloroform solutions at room temperature in the 10−5 M concentration range (Fig. 1). Compounds 3 and 5 were highly fluorescent in methanol. They exhibited photoluminescence properties and showed emission maxima at 373 and 413 nm (violet and violet-blue emission) upon excitation at 328 (Fig. 1(b) graph 3b) and 300 nm, respectively. Compounds 3 and 5 showed intense emission, which was slightly blue shifted when compared to the free ligand. The ligand exhibited emission maxima at 434 nm upon excitation at 300 nm. Compound 3 showed a blue shifted emission of 61 nm as it showed a maximum emission at 373 nm upon excitation at 300 nm and also showed an intense fluorescence emission at 367 nm upon excitation at 300 nm with a shift of 67 nm (Fig. 1(b)). Compounds 1, 2, 4 and 6 were relatively weak fluorescent materials and exhibited maxima at 350, 347, 347 and 348 nm respectively, upon excitation at the 300 nm. Compounds 1 and 4 showed strong blue shift emission when compared to the free ligand, probably due to the heavy metal effect or quenching behaviour or Ag⋯S/Hg⋯S secondary interactions. The violet and violet-blue emission of compounds 3 and 5 in the solution state implies that these compounds may be potentially applicable as materials for light emitting diode devices (LEDs).33,34
2.5 Thermal studies
The thermal properties of complexes 1–4 were studied by TG-DTA under a nitrogen atmosphere in the temperature range 25–900 °C at a controlled heating rate of 10 °C min−1 (Fig. 2). Thermogravimetric analysis of complex 1 showed it to be stable up to 210 °C and after that a single weight loss of 75.40% occurred in a single step between 210–320 °C to give silver sulfide (Ag2S) as the final residue (obs.: 21.43% and calc.: 22.75%).35 Complex 2 also showed almost the same pattern as that of complex 1. It was stable up to 213 °C and then lost its weight in a single step between 213–327 °C to give copper sulfide (CuS) as the end product (obs.: 21.85% and calc.: 23.19%).36 Complex 3 was stable up to 150 °C. Its low thermal stability was due to the presence of a chloroform molecule in its crystal lattice. Thus, it loses chloroform before it melts (309 °C) and showed a first step weight loss at 150–310 °C (weight loss calc.: 17.02 and obs.: 16.40%).10 Thus, heating at ∼200 °C may be a good method for the conversion of [Co(4-mpipdtc)3]·CHCl3 to [Co(4-mpipdtc)3]. Subsequently, in the second step between 310 and 355 °C it melts with decomposition to give cobalt sulfide (CoS2) as the final residue (obs.: 17.70% and calc.: 17.55%).12 Complex 4 completely decomposes in a three step process. It is stable up to 248 °C and the absence of any thermal change before this temperature indicated that sample restructuring does not take place before the degradation processes begins37 and then, decomposes in the first step up to 290 °C (weight loss calcd: 48.53 and observed: 49.70%), probably forming mercuric sulfide (HgS), which is also not a stable product. The HgS also decomposes rapidly and up to 500 °C and undergoes near complete vaporization.35 In the DTA curve, complex 4 showed two sharp endothermic curves around 250 °C near their melting temperatures. By comparing the TG and DTA curves, it was observed that the major weight loss process started after the melting temperatures of the compounds.38 The broad exothermic hump observed in complex 4 at 500 °C implies slow decomposition leading to volatilization upon heating (ESI Fig. 9 and 10†).37
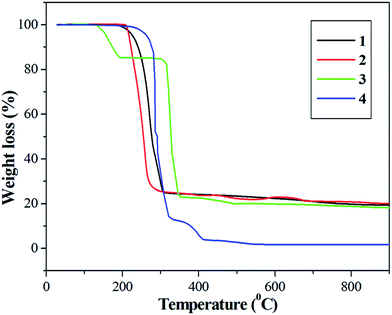 |
| | Fig. 2 TGA curves showing the thermal degradation of complexes 1–4 at a heating rate of 10 °C min−1 in a nitrogen atmosphere. | |
2.6 Electrochemical studies
The electrochemical behaviour of the ligand and its complexes were studied using cyclic voltammetry and are depicted as Fig. 3. The cyclic voltammograms were recorded with a 10−3 M concentration of the compounds at a platinum electrode using 0.05–0.1 M TBAP as supporting electrolyte in the potential range of −1.0 to +1.0 V with a scan rate of 20 mV s−1. The ligand and complexes 1, 3 and 4 showed irreversible behaviour while complex 2 showed reversible redox behaviour. Complex 2 may be applicable in fields such as electrochemical materials and electrochemical solar cells. The ligand showed cathodic and anodic peaks at 0.140 and −0.775 V, respectively, characterizing the irreversible redox behaviour (Fig. 3(a)). Complexes 1, 3 and 4 exhibited cathodic peaks at 0.080, 0.990 and 0.120 V and the corresponding anodic peaks at −0.260, 0.285 and −0.230 V, respectively. The separation between the cathodic and anodic peak potentials (DEp = Epa − Epc) of 0.340, 0.705 and 0.350 V for complexes 1, 3 and 4, respectively, indicate that these complexes show irreversible redox behaviour.39 Complex 2 exhibited the cathodic and anodic peaks at 0.545 and 0.485 V, respectively and the separation between the cathodic and anodic peak potentials (DEp = Epa − Epc) of 0.060 V indicates a reversible redox behaviour, which was assigned to the Cu(II)/Cu(I) couple with a formal redox potential E = (Epa + Epc)/2 = 0.515 V (Fig. 4).40 There was no shift in the peak potentials on scanning the voltammogram at different scan rates, which indicated that the complex exhibited completely reversible behaviour. Complex 2 also exhibited another cathodic and anodic peak at −0.380 and −0.520 V, respectively, due to the ligand moiety. The free ligand showed cathodic and anodic peaks at 0.140 and −0.775 V, respectively, which are shifted to −0.380 and −0.520 V due to formation of the complex.
 |
| | Fig. 3 The cyclic voltammograms obtained for the ligand and complexes: (a) the ligand in acetonitrile, (b) complex 1 in DCM-DMSO, (c) complex 2 in acetonitrile, (d) complex 3 in acetonitrile and (e) complex 4 in DMSO at Pt electrode using 0.1 M TBAP as the supporting electrolyte. | |
 |
| | Fig. 4 The cyclic voltammograms obtained for [Cu(mpipdtc)2] (2) at different scan rates. | |
2.7 Crystal structure description
The crystal structures of compounds 1–6 were obtained using single crystal X-ray diffraction data. The ORTEP diagrams of compounds 1–6 with atom numbering schemes are shown in Fig. 5–10. The details of the data collection, structure solution and refinement are listed in Table 2. Selected bond lengths and angles are included in Tables 3–8. Hydrogen bonding parameters for compounds 1–6 are given in Table 9.
Table 2 Crystallographic data, structure solution and refinement for compounds 1–6
| R1 = Σ||Fo| − |Fc||Σ|Fo|. R2 = [Σw (|Fo2| − |Fc2|)2/Σw|Fo2|2]1/2. |
| Parameters |
1 |
2 |
3 |
4 |
5 |
6 |
| Empirical formula |
C50H54Ag2N2P2S4 |
C14H24CuN2S4 |
C22H37Cl3CoN3S6 |
C13H17HgNS2 |
C14H24N2S4 |
C15H26N2S4 |
| Formula weight |
1088.87 |
412.13 |
701.18 |
451.99 |
348.59 |
362.62 |
| Crystal system |
Triclinic |
Monoclinic |
Triclinic |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
P |
P21/n |
P21/c |
P |
| T (K) |
293(2) |
120(2) |
120(2) |
293(2) |
120(2) |
293(2) |
| λ, Mo Kα (Å) |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
| a (Å) |
10.538(5) |
8.452(3) |
10.266(2) |
8.799(5) |
12.715(8) |
8.876(15) |
| b (Å) |
10.829(6) |
18.620(6) |
12.091(4) |
15.123(9) |
6.421(4) |
10.017(17) |
| c (Å) |
11.215(6) |
12.141(4) |
14.720(5) |
11.633(7) |
22.315(14) |
11.405(2) |
| α (°) |
78.16(5) |
90 |
113.48(3) |
90.00 |
90 |
87.26(15) |
| β (°) |
84.07(4) |
106.57(4) |
94.48(2) |
108.23(9) |
104.13(6) |
74.87(16) |
| γ (°) |
77.60(5) |
90 |
105.77(2) |
90.00 |
90 |
76.56(14) |
| V, (Å3) |
1221.2(11) |
1831.4(11) |
1576.3(9) |
1470.2(15) |
1767.1(2) |
952.0(3) |
| Z |
1 |
4 |
2 |
4 |
4 |
2 |
| ρcalcd (g cm−3) |
1.481 |
1.495 |
1.477 |
2.042 |
1.310 |
1.265 |
| μ (mm−1) |
1.074 |
5.890 |
10.462 |
10.732 |
4.867 |
0.495 |
| F (000) |
556 |
860 |
728 |
856 |
744 |
388 |
| Crystal size (mm3) |
0.30 × 0.27 × 0.23 |
0.30 × 0.27 × 0.23 |
0.55 × 0.25 × 0.21 |
0.30 × 0.24 × 0.21 |
0.49 × 0.31 × 0.04 |
0.30 × 0.27 × 0.23 |
| θ range for data collections (°) |
3.38–28.95 |
3.79–76.34 |
3.39–76.66 |
2.55–28.32 |
3.58–76.95 |
3.41–28.84 |
| Index ranges |
−13 ≤ h ≤ 14 |
−10 ≤ h ≤ 9 |
−12 ≤ h ≤ 7 |
−11 ≤ h ≤ 11 |
−15 ≤ h ≤ 15 |
−15 ≤ h ≤ 15 |
| −14 ≤ k ≤ 14 |
−20 ≤ k ≤ 23 |
−14 ≤ k ≤ 15 |
−20 ≤ k ≤ 20 |
−4 ≤ k ≤ 7 |
−4 ≤ k ≤ 7 |
| −15 ≤ l ≤ 15 |
−15 ≤ l ≤ 15 |
−18 ≤ l ≤ 18 |
−15 ≤ l ≤ 15 |
−28 ≤ l ≤ 27 |
−28 ≤ l ≤ 27 |
| No. of reflections collected |
6511 |
3796 |
6532 |
3666 |
3616 |
5001 |
| No. of independent reflections (Rint) |
4196 |
3578 |
6186 |
2245 |
2888 |
2469 |
| No. of data/restrains/parameters |
6511/0/271 |
3796/0/193 |
6532/0/320 |
3666/0/154 |
3616/15/215 |
5001/0/190 |
| Goodness-of-fit on F2 |
0.968 |
1.076 |
1.027 |
0.962 |
1.011 |
1.128 |
| R1a, wR2b [I > 2σ(I)] |
0.0445, 0.1118 |
0.0563, 0.1541 |
0.0471, 0.1269 |
0.0408, 0.0987 |
0.0508, 0.1305 |
0.0824, 0.2245 |
| R1a, wR2b (all data) |
0.0646, 0.1299 |
0.0582, 0.1570 |
0.0490, 0.1288 |
0.0679, 0.1111 |
0.0647, 0.1448 |
0.1115, 0.1695 |
| Largest difference in peak/hole (e Å−3) |
0.678, −0.696 |
1.458, −1.042 |
1.348, −0.861 |
1.480, −3.331 |
0.658, −0.558 |
0.475, −0.301 |
Table 3 The interatomic distances (Å) and angles (°) obtained for [Ag2(4-mpipdtc)2(PPh3)2] (1)
| Bond lengths (Å) |
Bond angles (°) |
| |
Exp. |
Cal. |
|
Exp. |
Cal. |
| Ag1–P1 |
2.427 |
2.550 |
S1i–Ag1–S2i |
67.143 |
65.1 |
| Ag1–S1 |
2.582 |
2.707 |
S1–Ag1i–S2 |
67.143 |
65.1 |
| Ag1–S2i |
2.587 |
2.678 |
S1–Ag1–S2i |
110.744 |
112.1 |
| Ag1–S1i |
2.787 |
2.678 |
S1–Ag1i–S1i |
105.023 |
112.5 |
| Ag1–Ag1i |
3.272 |
3.133 |
P1–Ag1–S1i |
117.073 |
113.5 |
| S2–C1 |
1.706 |
1.732 |
P1–Ag1–S1 |
120.353 |
118.3 |
| S1–C1 |
1.736 |
1.756 |
P1–Ag1–S2 |
123.334 |
123.3 |
| C1–N1 |
1.327 |
1.353 |
Ag1–S1–Ag1i |
74.983 |
67.5 |
Table 4 The interatomic distances (Å) and angles (°) obtained for [Cu(4-mpipdtc)2] (2)
| Bond lengths (Å) |
Bond angles (°) |
| |
Exp. |
Cal. |
|
Exp. |
Cal. |
| Cu–S1A |
2.295 |
2.398 |
S1B–Cu–S2B |
77.302 |
75.3 |
| Cu–S1B |
2.295 |
2.398 |
S1A–Cu–S2A |
77.592 |
75.3 |
| Cu–S2A |
2.305 |
2.398 |
S1B–Cu–S2A |
101.192 |
104.6 |
| Cu–S2B |
2.307 |
2.398 |
S1A–Cu–S2B |
103.502 |
104.6 |
| S1A–C1A |
1.729 |
1.738 |
C1A–S1A–Cu |
84.598 |
84.8 |
| S2A–C1A |
1.724 |
1.738 |
S2A–C1A–S1A |
113.201 |
179.9 |
| N1A–C1A |
1.325 |
1.339 |
S1A–Cu–S1B |
177.253 |
179.9 |
Table 5 The interatomic distances (Å) and angles (°) obtained for [Co(4-mpipdtc)3]·CHCl3 (3)
| Bond lengths (Å) |
Bond angles (°) |
| |
Exp. |
Cal. |
|
Exp. |
Cal. |
| Co–S1A |
2.264 |
2.331 |
S1A–Co–S2A |
76.562 |
75.9 |
| Co–S2A |
2.274 |
2.334 |
S2B–Co–S1B |
76.532 |
76.0 |
| Co–S1B |
2.278 |
2.331 |
S1C–Co–S2C |
76.182 |
75.9 |
| Co–S2B |
2.255 |
2.334 |
S1A–Co–S1B |
94.562 |
94.9 |
| Co–S1C |
2.261 |
2.331 |
S1C–Co–S1A |
94.873 |
94.8 |
| Co–S2C |
2.286 |
2.334 |
Cl2–C1S–Cl1 |
110.971 |
— |
| S1A–C1A |
1.719 |
1.728 |
S2B–Co–S1A |
167.413 |
167.3 |
| N1A–C1A |
1.321 |
1.344 |
S2A–C1A–S1A |
109.901 |
112.3 |
Table 6 The interatomic distances (Å) and angles (°) obtained for [PhHg(4-mpipdtc)] (4)
| Bond lengths (Å) |
Bond angles (°) |
| |
Exp. |
Cal. |
|
Exp. |
Cal. |
| Hg1–C4 |
2.059 |
2.237 |
C4–Hg1–S1 |
172.1 |
154.4 |
| Hg1–S1 |
2.393 |
2.692 |
C4–Hg1–S2 |
115.9 |
133.7 |
| Hg1–S2 |
2.854 |
2.846 |
S1–Hg1–S2 |
68.6 |
68.2 |
| S1–C7 |
1.751 |
1.747 |
C7–S1–Hg1 |
93.4 |
89.6 |
| S2–C7 |
1.720 |
1.753 |
C7–S2–Hg1 |
79.3 |
84.6 |
| N1–C7 |
1.318 |
1.341 |
S2–C7–S1 |
118.5 |
119.6 |
Table 7 The interatomic distances (Å) and angles (°) obtained for {4–mpipdtc2} (5)
| Bond lengths (Å) |
Bond angles (°) |
| |
Exp. |
Cal. |
|
Exp. |
Cal. |
| S1A–C1A |
1.827 |
1.860 |
C1A–S1A–S1B |
102.09 |
104.5 |
| S2A–C1A |
1.657 |
1.659 |
N1A–C1A–S2A |
126.02 |
126.0 |
| C1A–N1A |
1.341 |
1.351 |
N1A–C1A–S1A |
112.22 |
111.5 |
| S1A–S1B |
2.008 |
2.037 |
S2A–C1A–S1A |
121.70 |
122.3 |
Table 8 The interatomic distances (Å) and angles (°) obtained for {CH24-mpipdtc2} (6)
| Bond lengths (Å) |
Bond angles (°) |
| |
Exp. |
Cal. |
|
Exp. |
Cal. |
| S1–C7 |
1.7907 |
1.812 |
C7–S1–C15 |
102.34 |
102.8 |
| S3–C14 |
1.783 |
1.812 |
C14–S3–C15 |
102.14 |
102.8 |
| S2–C7 |
1.656 |
1.678 |
S3–C15–S1 |
116.84 |
116.5 |
| S4–C14 |
1.659 |
1.678 |
S2–C7–S1 |
121.54 |
122.5 |
| S1–C15 |
1.796 |
1.820 |
S4–C14–S3 |
122.04 |
122.5 |
| S3–C15 |
1.791 |
1.819 |
N1–C7–S2 |
125.15 |
124.3 |
| N1–C7 |
1.333 |
1.354 |
C4–N1–C6 |
110.56 |
111.9 |
| N2–C14 |
1.342 |
1.354 |
C7–N1–C6 |
122.26 |
122.1 |
Table 9 The hydrogen bond parameters [(Å) and (°)], C–H⋯π and S⋯S interactions obtained for compounds 1–6
| Compounds |
D–H⋯A |
dD–H |
dH⋯A |
dD⋯A |
<DHA |
Symmetry equivalent operators |
| 1 |
C11–H11⋯S11 |
0.930 |
2.786 |
3.696 |
166.17 |
2 – x, 1 − y, –z |
| C6–H6B⋯Cg |
0.970 |
2.775 |
3.646 |
149.80 |
−x, −y, −z |
| 2 |
C6B–H6BA⋯S2B |
0.991 |
2.911 |
3.653 |
132.43 |
x, 0.5 − y, 0.5 + z |
| S1A⋯S1A |
3.473 |
| 3 |
C4A–H4AA⋯S2C |
1.000 |
2.994 |
3.895 |
150.52 |
2 − x, 1 − y, 1 − z |
| C6A–H6AA⋯S2C |
0.990 |
2.920 |
3.827 |
151.84 |
2 − x, 1 − y, 1 − z |
| 4 |
C8–H3B⋯Cg |
0.970 |
2.920 |
3.692 |
137.32 |
0.5 − x, 0.5 + y, 0.5 − z |
| 5 |
C6A–H6AB···S2A |
0.990 |
2.806 |
3.719 |
153.68 |
1 − x, 0.5 + y, 1.5 − z |
| S1A⋯S2A |
3.547 |
| 6 |
C4–H4B⋯S4 |
0.970 |
2.791 |
3.674 |
151.69 |
2 − x, 2 − y, 1 − z |
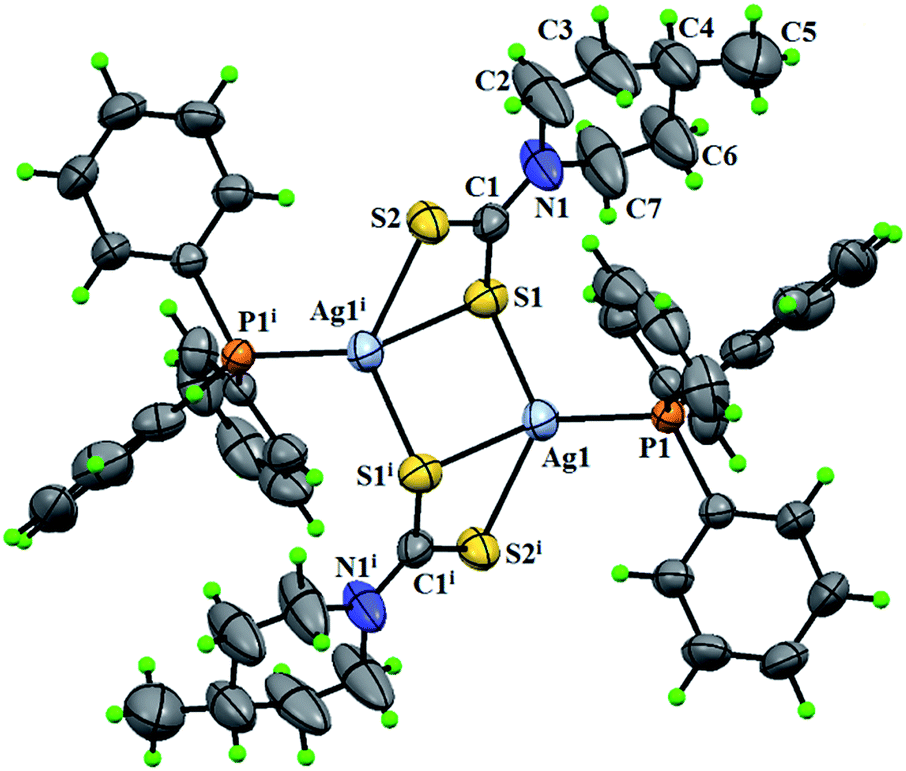
2.7.1 Crystal structure description of [Ag2(4-mpipdtc)2(PPh3)2] (1). Fig. 5 shows ORTEP diagram of complex 1 along with the atom numbering scheme. Complex 1 forms a dinuclear molecule in which each silver ion is bonded through two bridging uninegative bidentate dithiocarbamato ligands via sulfur and one triphenylphosphine co-ligand. The ligand dithiocarbamato shows a mixed structural function that includes formation of chelate rings and the sulfur bridges in the dimeric Ag(I) complex 1. The Ag–S bond lengths at the sulfur bridge (Ag(1)–S(1) = 2.582(11) Å) are shorter than the non-bridging Ag–S bond distances (Ag(1)–S(1) = 2.787(12) and Ag(1)–S(2) = 2.587(12) Å) indicating that the bridging Ag–S bonds are stronger than the non-bridging Ag–S bonds. The ligand having mixed structural function coordinates to Ag(I) ion and forms tricyclic fragments [Ag2S4C2]. The geometry of this [Ag2S4C2] ring can be approximated to a chair conformation.41 The bridging bond angles with Ag and S atoms of different ligand units (S(1)–Ag(1)–S(2) = 110.74(4)°) are slightly greater than a normal tetrahedral bond angle of 109.47°. The bond length P(1)–Ag = 2.4275(9) Å is quite similar to the reported Ag(I) complexes of triphenylphosphine.42 The carbon–nitrogen bond length of 1.327(6) Å in the carbodithioate moiety indicates a substantial delocalization of the π-electron density attributing some double bond character.30 The crystal structure of the complex is stabilized by weak intermolecular C–H⋯S interactions occurring between the dithio sulfur and C–H hydrogen of the phenyl ring (ESI Fig. 11†) (Table 9). In addition, the structure is stabilized by intramolecular C–H⋯π interactions (2.775 Å) between the C–H of the piperidine ring and π electrons of a phenyl ring of PPh3, which is well within the reported range (2.75–2.89 Å) (ESI Fig. 12†).43 The intermolecular interactions are clearly hydrogen bond interactions while the intramolecular interactions deal with the C–H⋯π interactions only.
 |
| | Fig. 5 The ORTEP diagram of [Ag2(4-mpipdtc)2(PPh3)2] (1). | |
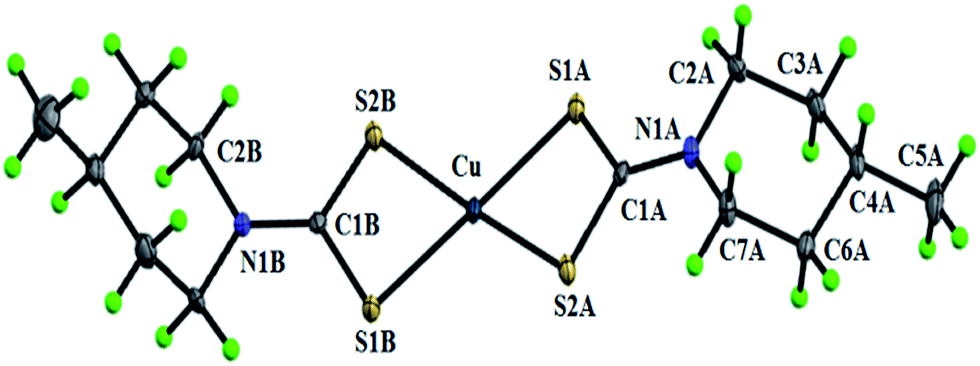
2.7.2 Crystal structure description of [Cu(4-mpipdtc)2] (2). Fig. 6 shows the ORTEP diagram of complex 2 with the atom numbering scheme. In complex 2 the copper(II) ion is bonded through four sulfur atoms of two dithiocarbamato ligands. The CuS4 core exhibits a distorted square planar geometry. The four Cu–S bond lengths are unequal and lie in the range 2.295(6)–2.307(6) Å. The bite angles of two chelate rings are slightly different {S(1A)–Cu–S(2A) = 77.59(2) and S(1B)–Cu–S(2B) = 77.30(2)°} and less than normal bond angle (90°), which indicate that the complex has a distorted square planar geometry. The other S(1A)–Cu–S(2B) and S(1B)–Cu–S(2A) bond angles are 103.50(2) and 101.19(2)°, respectively. The N–C bond lengths of the two NCS2 moieties {N(1A)–C(1A) = 1.325(3) and N(1B)–C(1B) = 1.321(3) Å} are shorter than a single N–C bond length attributed to partial double bond character, which suggests the delocalization of the π-electrons in the NCS2 moiety.30 The crystal structure of complex 2 is stabilized by weak intermolecular C–H⋯S interactions occurring between the dithiosulfur atom and C–H hydrogen in the piperidine ring (ESI Fig. 13†) (Table 9). The crystal structure is further stabilized by S⋯S interactions, which lead to a wave-like architecture (ESI Fig. 14†).
 |
| | Fig. 6 The ORTEP diagram of [Cu(4-mpipdtc)2] (2). | |
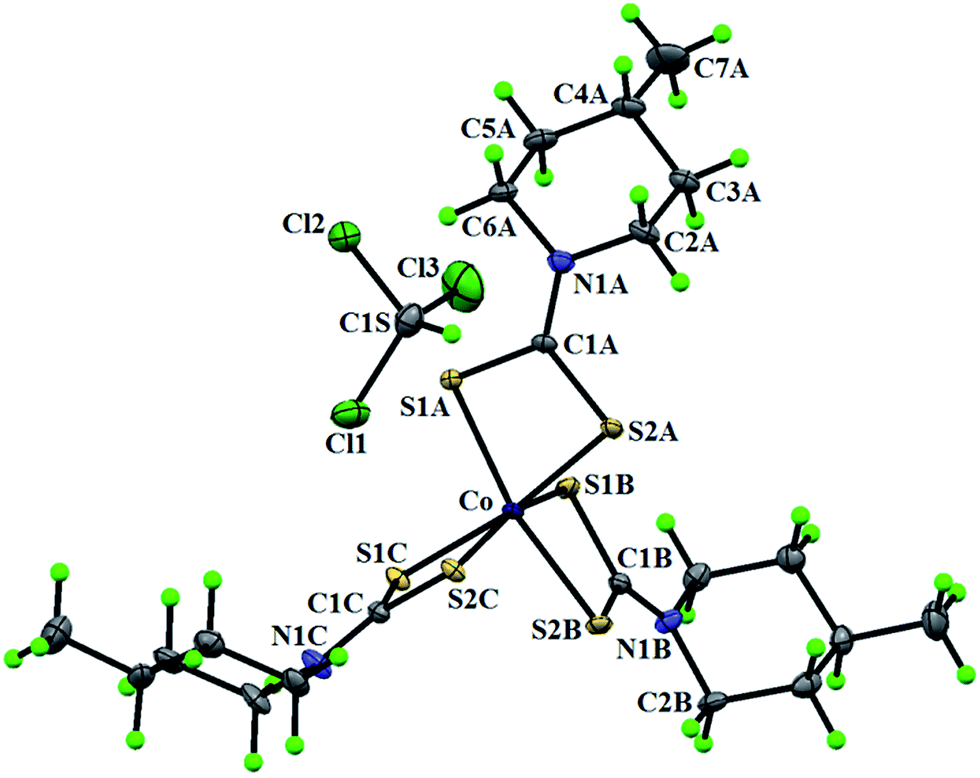
2.7.3 Crystal structure description of [Co(4-mpipdtc)3]·CHCl3 (3). Fig. 7 shows the ORTEP diagram of complex 3 along with the atom numbering scheme. The coordination sphere of complex 3 is fulfilled by six dithio-sulfur atoms from three bidentate dithiocarbamato moieties of the ligand. The formation of three four membered CS2Co chelate rings, with a bite angle in the range of 76.18(2)–76.56(2)° represent a minor deviation from an ideal octahedral geometry. The Co–S bond distances are unequal being 2.264(6), 2.274(7), 2.278(7), 2.255(6), 2.261(7) and 2.286(7) Å and are comparable to the bond distances reported earlier for other similar cobalt dithio complexes.44–46 The carbon–sulfur distances within the chelate rings are intermediate between single and double bond lengths. These bond lengths, C(1A)–S(1A) = 1.719(3), C(1A)–S(2A) = 1.716(2), C(1B)–S(1B) = 1.714(2), C(1B)–S(2B) = 1.714(3), C(1C)–S(1C) = 1.713(3) and C(1C)–S(2C) = 1.715(2) Å (Table 5) suggest a considerable delocalization of charge in the chelate ring.46 In the solid state, complex 3 is stabilized via two types of intermolecular C–H⋯S interactions that occur between the dithio sulfur atoms and the hydrogen atoms in the piperidine ring/hydrogen atoms of chloroform molecules (Table 9) (ESI Fig. 15†).
 |
| | Fig. 7 The ORTEP diagram of [Co(4-mpipdtc)3]·CHCl3 (3). | |
2.7.4 Crystal structure description of [PhHg(4-mpipdtc)] (4). Fig. 8 shows the ORTEP diagram of complex 4 together with the atom numbering scheme. Hg(II) is bonded with phenyl carbon and the dithiosulfur atoms of the ligand, and the complex has an almost linear geometry. The C–S bond distances of 1.751(6) and 1.720(6) Å in complex 4 agree well with those reported for other similar complexes,47,48 being an intermediate between the C–S single (1.82 Å) and CS double (1.56 Å) bond distances.49 The Hg–S1 bond length is 2.393(2) Å, whilst that of Hg(1)–S(2) is 2.854(2) Å, which suggests that the bond between Hg and S(2) is longer than Hg(1)–S(1), thereby reflecting the propensity of a linear geometry around Hg(II) ion. The bond angle of 172.18(16)° indicates a deviation from the ideal linear geometry. The intermolecular distance between Hg(II) ion and S(2) is 3.166 Å, which suggests a strong interaction between metal ion and sulfur atom. In the solid state, the crystal structure is stabilized by C–H⋯π interactions (2.920 Å) occurring between C–H of piperidine and π electrons of the phenyl rings (ESI Fig. 16†).
 |
| | Fig. 8 The ORTEP diagram of [PhHg(4-mpipdtc)] (4). | |
2.7.5 Crystal structure description of (4-mpipdtc)2 (5). Fig. 9 shows the ORTEP diagram of compound 5 along with the atom numbering scheme. The bond lengths and angles are slightly different in the two halves of (4-mpipdtc)2 (5) with an S(2A)–S(1A)–S(1B)–S(2B) torsion angle of 90.20°, which indicates that they are nearly perpendicular to each other. In addition, the S(1)–S(2) bond length of 2.0081(10) Å is found in the normal range for a disulfide bond. The C(1A)–S(1A), C(1B)–S(1B) and S(1A)–S1(B) bond lengths of 1.827(3), 1.832(3) and 2.0081(10) Å, respectively show C–S and S–S single bond character and indicate that compound 5 is present in the disulfide form.20 This is further supported by the adjacent C(1A)–S(2A) and C(1B)–S(2)B bond lengths of 1.657(3) and 1.646(3) Å, which are shorter than those in complexes 1–4 (Tables 3–7), thus suggesting double bond character. The crystal structure is stabilized by intermolecular C–H⋯S hydrogen bonding and S⋯S interactions, which leads to a linear chain architecture (ESI Fig. 17†) (Table 9).
 |
| | Fig. 9 The ORTEP diagram of (4-mpipdtc)2 (5). | |
2.7.6 Crystal structure description of {CH2(4-mpipdtc)2} (6). Fig. 10 shows the ORTEP diagram of {CH2(4-mpipdtc)2} (6) with the atom numbering scheme. Crystal structure of compound 6 indicates that the methylene group is bonded through the two sulfur atoms of two units of dithiocarbamate ligand at distances of 1.796(7) Å {C(15)–S(1)} and 1.791(7) Å {C(15)–S(3)}. The bond angle of 116.8(4)° {S(1)–C(15)–S(3)} is larger than the ideal tetrahedral value of 109.47°, which may be due to repulsion between the two CS bonding electron pairs. The bonds C(7)–N(1) {1.333(8) Å} and C(14)–N(2) {1.342(8) Å} are shorter than the reported C–N average bond lengths (1.462 Å).22 The averages of the C–S and CS bond distances are 1.790 and 1.657 Å, respectively, which are well within the range of the reported single and double bond lengths.17,22 In the solid state, the crystal structure is stabilised by weak intermolecular C–H⋯S interactions occurring between hydrogens in the piperidine ring and sulfur atom in the dithiocarbamate ligand (ESI Fig. 18†) (Table 9).
 |
| | Fig. 10 The ORTEP diagram of CH2(4-mpipdtc)2 (6). | |
2.8 Frontier molecular orbital analysis
The HOMO and LUMO are very important parameters for quantum chemistry. The most important frontier molecular orbitals (FMOs) such as the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) play a crucial part in the chemical stability of a molecule.50 The HOMO represents the ability to donate an electron and LUMO as an electron acceptor represents the ability to accept an electron. The energy gap between the HOMO and LUMO also determines the chemical reactivity, optical polarizability and chemical hardness-softness of a molecule.51 HOMO and LUMO analysis is also used to determine the charge transfer within a molecule. Considering the chemical hardness, if a molecule has a large HOMO–LUMO gap, it is a hard molecule or small HOMO–LUMO gap it is a soft molecule. One can also relate the stability of a molecule to hardness, which means that the molecule with the least HOMO–LUMO gap is more reactive. Soft systems are large and highly polarizable, while hard systems are relatively small and much less polarisable.
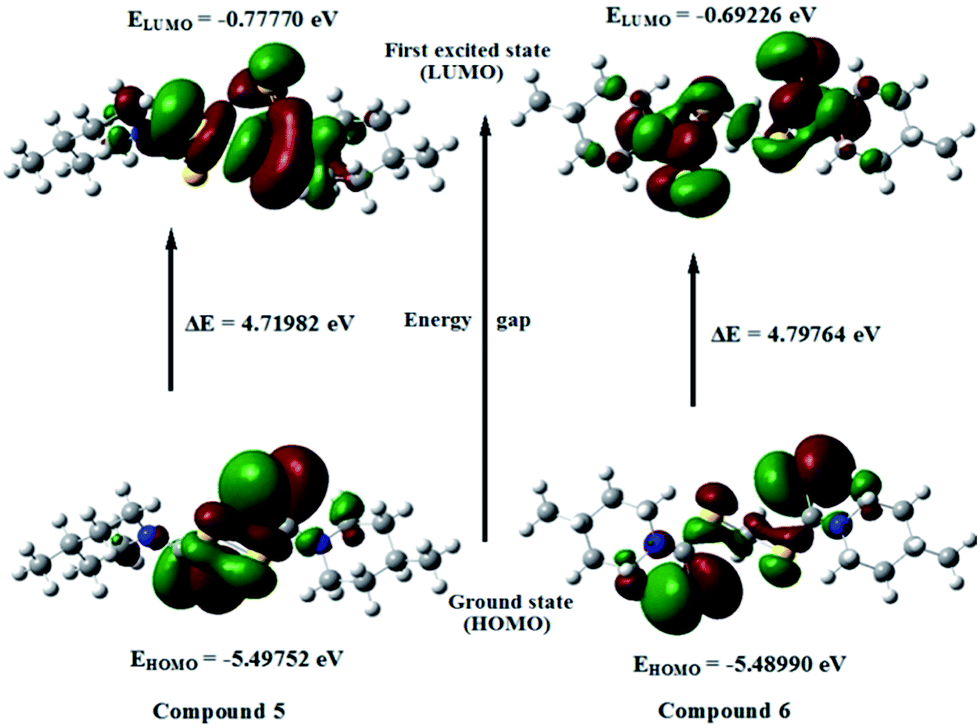
The analysis of the wavefunction indicates that the electron absorption corresponds to the transition from the ground state to the first excited state and is mainly described by one-electron excitation from the HOMO to LUMO.52 Fig. 11 and 12 show the shapes and energy levels of the HOMO and LUMO for complexes 1–6. The HOMO of complexes 1, 2 and 3 show that the electron density is mainly localized on the chelate rings while the LUMO of complex 1 shows that the electron density would be localized over the piperidine ring. The LUMO of complexes 2 and 3 show that the electron density would be localized over piperidine ring and chelate ring. The HOMO of complex 4 shows that electron density is localized on the dithio sulfur atoms and bonded mercury(II) ion whereas the LUMO of complex 4 shows that the electron density would be localized over the piperidine ring as well as the mercury(II) ion and bonded dithio sulfur atoms. The HOMO of compound 5 shows that the electron density is mainly localized on the disulfide sulfur atoms while the LUMO of compound 5 shows that the electron density is going to be localized over piperidine ring as well as the disulfide sulfur atoms. The HOMO of compound 6 shows that electron density is localized on the both dithio sulfur, methylene sulfur atoms and piperidine nitrogen atom, whereas LUMO of compound 6 shows that the electron density would be localized over both dithio sulfur, methylene sulfur atoms and piperidine ring. The HOMO–LUMO energy gaps for complexes 1–4 are 3.94946, 4.20471, 3.96279 and 4.55900 eV, respectively. It is pertinent to mention here that the energy gap is smaller in complexes 1 and 3 than in complexes 2 and 4. Therefore, complexes 1 and 3 can serve as better targets for photophysical studies and electronics applications when compared to complexes 2 and 4.53,54 In addition, complexes 1 and 3 are softer and hence more reactive when compared to complexes 2 and 4. The HOMO–LUMO energy gaps for compounds 5 and 6 are 4.71982 and 4.79764 eV, respectively (Table 10). The electronic transition from the ground state to the first excited state for these molecules is due to the transfer of electrons from the HOMO to LUMO level being mainly the π⋯π* type.
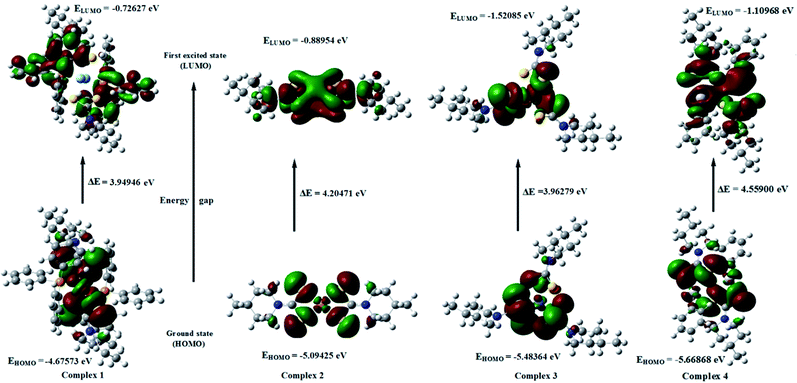 |
| | Fig. 11 The frontier molecular orbitals of complexes 1, 2, 3 and 4. | |
 |
| | Fig. 12 The frontier molecular orbitals of molecules 5 and 6. | |
Table 10 The calculated frontier molecular orbital energies (eV) obtained for compounds 1–6
| Molecules |
Energies (eV) |
| HOMO |
LUMO |
Energy gap |
| 1 |
−4.67573 |
−0.72627 |
3.94946 |
| 2 |
−5.09425 |
−0.88954 |
4.20471 |
| 3 |
−5.48364 |
−1.52085 |
3.96279 |
| 4 |
−5.66868 |
−1.10968 |
4.55900 |
| 5 |
−5.49752 |
−0.77770 |
4.71982 |
| 6 |
−5.48990 |
−0.69226 |
4.79764 |
3. Conclusions
In conclusion, we report the syntheses, structural investigation and physicochemical properties (photoluminescence, thermal analyses and cyclic voltammetry) of Ag(I), Cu(II), Co(III) and Hg(II) complexes bearing the 4-methyl-piperidine-carbodithioate ligand. Additionally, Mn(II) assisted formation of a disulfide compound and the Ag(I) mediated synthesis of a carbothioylsulfanyl derivative have also been discovered. The solid state structures of the Ag, Cu, Co complexes and the disulfide and carbothioylsulfanyl derivatives revealed intermolecular C–H⋯S interactions including S⋯S interactions in the Cu complex and the disulfide derivative. Further, intramolecular C–H⋯π and the Hg⋯S secondary interactions were found in the Hg(II) complex. The photoluminescence properties of the Co(III) complex and the disulfide derivative make them potential materials to be useful in light emitting diodes (LED). When compared to the free ligand, a large blue shift emission in its Ag(I) and Hg(II) complexes was a consequence of the quenching behaviour and/or an effect of the Hg⋯S/Ag⋯S secondary interactions. The reversible redox behaviour of the Cu(II) complex makes it a potential probe in electrochemical materials and electrochemical solar cells. The thermal degradation of the metal complexes, as analysed by TG-DTA, showed strong evidence for the formation of their respective metal sulfides as the final thermally stable chemical entities.
4. Experimental
4.1 Materials and methods
4-Methyl-piperidine was purchased from Sigma Aldrich. Other chemicals were of reagent grade and used as purchased without further purification. All synthetic manipulations were carried out open to the ambient atmosphere and at room temperature. The solvents were distilled before use following standard procedures. The carbon, hydrogen, nitrogen and sulfur contents were determined on a CHN Model CE-440 Analyzer and on an Elementar Vario EL III Carlo Erba 1108. Infrared spectra were recorded in the 4000–400 cm−1 region as KBr pellets on a Varian Excalibur 3100 FT-IR spectrophotometer. 1H and 13C NMR spectra were recorded in CDCl3 on a JEOL AL 300 FT NMR spectrometer using TMS as an internal reference. The fluorescent data were collected at room temperature on a Varian CARYECLIPSE spectrophotometer in CHCl3 (1 and 4) and MeOH (2, 3, 5 and 6). Thermogravimetric analysis (TG-DTA) of complexes 1–2 was performed using a Perkin Elmer-STA 6000 thermal analyzer, TA Instrument and complexes 3–4 were performed using a thermal analyzer Model No-TGA/DSC1 STAR System Germany, in a nitrogen atmosphere at a heating rate of 10 °C min−1.
4.2 Cyclic voltammetry
Electrochemical studies were performed on a Metrohm (Netherlands) Instrument (Autolab PGSTAT204) potentiostat/galvanostat attached with NOVA 1.11 software. A three electrode, one compartment cell with solid platinum electrode as the working electrode, platinum wire as the counter electrode and Ag/AgCl as the reference electrode was used. All electrochemical studies were carried out at room temperature under a nitrogen atmosphere.
4.3 X-ray crystallography
Structural measurements of compounds 1–6 were performed on a computer-controlled Oxford Gemini diffractometer equipped with a CrysAlis CCD software using a graphite mono-chromated Mo Kα (λ = 0.71073 Å) radiation source at 293 K. Multi-scan absorption correction was applied to the X-ray data collection for all the compounds. The structures were solved using direct methods (SHELXL-2013) and refined against all data by full matrix least-square on F2 using anisotropic displacement parameters for all non-hydrogen atoms. All hydrogen atoms were included in the refinement at geometrically ideal positions and refined with the Riding model.55 The MERCURY package and ORTEP-3 for Windows program were used for generating the molecular graphics.56,57
4.4 Quantum chemical calculations
To understand the binding sites and to verify the composition of the complexes, density functional theory (DFT) calculations were performed. In these calculations, the B3LYP density functional theory method was used, which is a hybrid version of the DFT and Hartree–Fock (HF) methods.58 In this method, the exchange energy from Becke's exchange functional was combined with the exact energy obtained from the Hartree–Fock theory.59 The three parameters define the hybrid functional, specifying how much of the exact exchange is mixed in along with the component exchange and correlation functionals.60 The 6-31G** basis set for all the atoms except metal ions was used. The LanL2DZ basis set with an effective core pseudo potential was used for the metal atoms.61 All the geometry optimizations and frequency calculations (to verify a genuine minimum energy structure) were performed using the Gaussian 09 program package.62 It worth pointing out here that the starting geometries were chosen based on the X-ray crystallographic structures with required modification for the computations if needed.
4.5 Synthesis
4.5.1 Synthesis of potassium 4-methyl-piperidine-carbodithioate {K+(4-mpipdtc)−}. Potassium 4-methyl-piperidine-carbodithioate was prepared by the dropwise addition of carbon disulfide (1.5 mL, 20 mmol) to an ethanolic solution of 4-methyl-piperidine (2.4 mL, 20 mmol) and potassium hydroxide (1.2 g, 20 mmol). The reaction mixture was stirred continuously for 2 h. The solid potassium 4-methyl-piperidine-carbodithioate {K+(4-mpipdtc)−} obtained was filtered, washed with ethanol and dried under reduced pressure (Scheme 2). Yield: 75%; mp 185 °C. Anal. calc. for C7H12NS2K (213.40): C, 35.78; H, 5.66; N, 6.56; S, 30.05. Found: C, 35.95; H, 5.90; N, 6.30; S, 29.70%. IR (KBr, cm−1): 2951 ν(C–H), 1585 ν(C–N), 1015 ν(CS). 1H NMR (δ, ppm): 0.95 (d, 3H, CH3), 1.20–1.59 (m, 4H, H2), 1.70–1.75 (m, 1H, H3), 3.76 (t, 2H, H1-axial), 5.67 (d, 2H, H1-equatorial). 13C NMR (δ, ppm): 21.56 (CH3), 30.89 (C2), 34.35 (C3), 51.29 (C1), 208.76 (CS) (Scheme 4). UV-Vis. (CHCl3, λmax/nm, ε/M−1 cm−1): 265 (1.2 × 104), 297 (1.2 × 104).
4.5.2 Synthesis of [Ag2(4-mpipdtc)2(PPh3)2] (1). AgNO3 (0.169 g, 1 mmol) was added to a 10 mL methanol solution of {K+(4-mpipdtc)−} (0.213 g, 1 mmol) and stirred for 1 h at room temperature. A white precipitate was obtained. Triphenylphosphine (0.262 g, 1 mmol) solution in chloroform was added to the above suspension. A colourless clear solution was obtained, which was filtered and kept for crystallization. Colourless crystals suitable for X-ray analysis were obtained after the slow evaporation of the solvent over a period of 15 days (Scheme 2). Yield: 58%; mp 196 °C. Anal. calc. for C50H54Ag2N2P2S4 (1088.87): C, 55.14; H, 4.96; N, 2.57; S, 11.76. Found: C, 55.00; H, 5.20; N, 2.40; S, 11.52%. IR (KBr, cm−1): 3058 ν(C–H, phenyl) (calc. 3177), 2989 ν(C–H) (calc. 2948), 1469 ν(C–N) (calc. 1462), 962 ν(CS) (calc. 960), ν(Ag–S) (calc. 320). 1H NMR (δ, ppm): 0.93 (d, 3H, CH3), 1.32–1.57 (m, 4H, H2), 1.68 (m, 1H, H3), 3.07 (t, 2H, H1-axial), 5.30 (d, 2H, H1-equatorial), 7.34–7.49 (m, 15H, phenyl H). 13C NMR (δ, ppm): 21.27 (CH3), 30.39 (C2), 33.87 (C3), 52.74 (C1), 126.21–134.09, 207.17 (CS) (Scheme 4). UV-Vis. (CHCl3, λmax/nm, ε/M−1 cm−1): 263 (9 × 103).
4.5.3 Synthesis of [Cu(4-mpipdtc)2] (2). Cu(OAc)2·H2O (0.200 g, 1 mmol) was added to a solution of {K+(4-mpipdtc)−} (0.426 g, 1 mmol) in 20 mL of a methanol-chloroform mixture and stirred for 3 h at room temperature. A clear green solution was obtained, which was filtered and kept for crystallization. Prism shaped dark blue crystals suitable for X-ray analysis were obtained over a period of 10 days. Yield: 78%; mp 213 °C. Anal. calc. for C14H24CuN2S4 (412.13): C, 40.79; H, 5.86; N, 6.80; S, 31.04. Found: C, 40.25; H, 5.60; N, 6.75; S, 31.33%. IR (KBr, cm−1): 2948 ν(C–H) (calc. 2995), 1504 ν(C–N) (calc. 1492), 965 ν(CS) (calc. 966), 429 ν(Cu–S) (calc. 405). UV-Vis. (MeOH, λmax/nm, ε/M−1 cm−1): 272 (8 × 103), 439 (3 × 102).
4.5.4 Synthesis of [Co(4-mpipdtc)3]·CHCl3 (3). CoCl2 (0.130 g, 1 mmol) was added to a solution of {K+(4-mpipdtc)−} (0.639 g, 3 mmol) in 20 mL of a methanol-chloroform mixture and stirred for 3 h at room temperature. A clear green solution was obtained, which was filtered and kept for crystallization. Dark green crystals suitable for X-ray analysis were obtained over a period of 10 days (Scheme 2). Yield: 80%; mp 309 °C. Anal. calc. for C22H37Cl3CoN3S6 (701.18): C, 37.68; H, 5.31; N, 5.60; S, 18.28. Found: C, 37.88; H, 5.15; N, 5.40; S, 18.53%. IR (KBr, cm−1): 2949 ν(C–H) (calc. 2993), 1493 ν(C–N) (calc. 1493), 966 ν(CS) (calc. 875), 428 ν(Co–S) (calc. 358). 1H NMR (δ, ppm): 0.96 (d, 3H, CH3), 1.23 (m, 4H, H2), 1.71 (m, 1H, H3), 2.89 (t, 2H, H1-axial), 4.66 (d, 2H, H1-equatorial). 13C NMR (δ, ppm): 21.32 (CH3), 30.65 (C2), 33.13 (C3), 45.48 (C1), 203.55 (CS) (Scheme 4). UV-Vis. (MeOH, λmax/nm, ε/M−1 cm−1): 271 (3 × 104), 328 (2 × 104).
4.5.5 Synthesis of [PhHg(4-mpipdtc)] (4). A methanol-chloroform solution of PhHg(OAc) (0.337 g, 1 mmol) was added to a 20 mL of a methanol-chloroform mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) containing {K+(4-mpipdtc)−} (0.213 g, 1 mmol) and stirred for 3 h at room temperature. A colourless clear solution was obtained, which was filtered and kept for crystallization. Rod shaped crystals suitable for X-ray analyses were obtained over a period of 7 days (Scheme 2). Yield: 75%; mp 248 °C. Anal. calc. for C13H17HgNS2 (451.99): C, 34.54; H, 3.78; N, 3.10; S, 14.18. Found: C, 34.35; H, 3.60; N, 3.01; S, 14.33%. IR (KBr, cm−1): 3062 ν(C–H phenyl) (calc. 3120), 2947 ν(C–H) (calc. 2999), 1484 ν(C–N) (calc. 1484), 968 ν(CS) (calc. 960), 430 ν(Hg–S) (calc. 428). 1H NMR (δ, ppm): 0.93 (d, 3H, CH3), 1.32–1.57 (m, 4H, H2), 1.68 (m, 1H, H3), 3.10 (t, 2H, H1-axial), 4.95 (d, 2H, H1-equatorial), 7.34–7.49 (m, 5H, phenyl H). 13C NMR (δ, ppm): 21.19 (CH3), 30.25 (C2), 33.79 (C3), 52.88 (C1), 128.23–154.46, 200.81 (CS) (Scheme 4). UV-Vis. (CHCl3, λmax/nm, ε/M−1 cm−1): 295 (5 × 103).
:1) containing {K+(4-mpipdtc)−} (0.213 g, 1 mmol) and stirred for 3 h at room temperature. A colourless clear solution was obtained, which was filtered and kept for crystallization. Rod shaped crystals suitable for X-ray analyses were obtained over a period of 7 days (Scheme 2). Yield: 75%; mp 248 °C. Anal. calc. for C13H17HgNS2 (451.99): C, 34.54; H, 3.78; N, 3.10; S, 14.18. Found: C, 34.35; H, 3.60; N, 3.01; S, 14.33%. IR (KBr, cm−1): 3062 ν(C–H phenyl) (calc. 3120), 2947 ν(C–H) (calc. 2999), 1484 ν(C–N) (calc. 1484), 968 ν(CS) (calc. 960), 430 ν(Hg–S) (calc. 428). 1H NMR (δ, ppm): 0.93 (d, 3H, CH3), 1.32–1.57 (m, 4H, H2), 1.68 (m, 1H, H3), 3.10 (t, 2H, H1-axial), 4.95 (d, 2H, H1-equatorial), 7.34–7.49 (m, 5H, phenyl H). 13C NMR (δ, ppm): 21.19 (CH3), 30.25 (C2), 33.79 (C3), 52.88 (C1), 128.23–154.46, 200.81 (CS) (Scheme 4). UV-Vis. (CHCl3, λmax/nm, ε/M−1 cm−1): 295 (5 × 103).
4.5.6 Synthesis of (4-mpipdtc)2 (5). Mn(OAc)2·4H2O (0.245 g, 1 mmol) was added to a solution of {K+(4-mpipdtc)−} (0.426 g, 2 mmol) in 20 mL of methanol and stirred for 3 h at room temperature. A clear yellow solution was obtained, which was filtered and kept for crystallization. Rod-shaped crystals suitable for X-ray analysis were obtained over a period of 15 days (Scheme 3). Yield: 82%; mp 102 °C. Anal. calc. for C14H24N2S4 (348.59): C, 48.23; H, 5.31; N, 6.93; S, 36.78. Found: C, 48.43; H, 5.23; N, 6.76; S, 36.58%. IR (KBr, cm−1): 2950 ν(C–H) (calc. 2966), 1481 ν(C–N) (calc. 1479), 959 ν(CS) (calc. 902), 816 ν(C–S) (calc. 821), 556 ν(S–S) (calc. 564). 1H NMR (δ, ppm): 0.99 (d, 3H, CH3), 1.39–1.57 (m, 4H, H2), 1.79 (m, 1H, H3), 3.32 (t, 2H, H1-axial), 5.00 (d, 2H, H1-equatorial). 13C NMR (δ, ppm): 21.20 (CH3), 30.85 (C2), 33.83 (C3), 47.43 (C1), 192.82 (CS2) (Scheme 4). UV-Vis. (MeOH, λmax/nm, ε/M−1 cm−1): 283 (4 × 104).
4.5.7 Synthesis of CH2(4-mpipdtc)2 (6). AgNO3 (0.169 g, 1 mmol) was added to a solution of {K+(4-mpipdtc)−} (0.426 g, 2 mmol) in 20 mL of methanol and stirred for 1 h at room temperature. A white precipitates was obtained, to which dichloromethane (10 mL) was added dropwise and stirred for 2 h, followed by filtration and kept for crystallization. Rod-shaped colourless crystals of 6 suitable for X-ray analysis were obtained over a period of 15 days (Scheme 3). Yield: 80%; mp 140 °C. Anal. calc. for C15H26N2S4 (362.62): C, 49.68; H, 7.22; N, 7.72; S, 35.36. Found: C, 49.42; H, 7.23; N, 7.81; S, 35.53%. IR (KBr, cm−1): 2924 ν(C–H), 1434 ν(C–N), 998 ν(CS), 714 ν(C–S). 1H NMR (δ, ppm): 0.95 (d, 3H, CH3), 1.24–1.43 (m, 4H, H2), 1.65 (m, 1H, H3), 2.95 (t, 2H, H1-axial), 4.09 (d, 2H, H1-equatorial), 5.37 (s, 2H, SCH2), 13C NMR (δ, ppm): 21.19 (CH3), 30.83 (C2), 34.00 (C3), 52.04 (C1), 54.52 (SCH2S) 193.62 (CS2), UV-Vis. (MeOH, λmax/nm, ε/M−1 cm−1): 260 (1 × 104).
Acknowledgements
Paras Nath thanks the UGC, India for the award of JRF and Dr M. K. Bharty is thankful to the Science and Engineering Research Board, India for the award of a Project (No. SB/EMEQ-150/2014; Diary No. SERB/F/372/2015-16).
Notes and references
-
(a) G. Hogarth, Prog. Inorg. Chem., 2005, 53, 71–561 CrossRef CAS;
(b) E. R. T. Tiekink and I. Haiduc, Prog. Inorg. Chem., 2005, 54, 127–319 CrossRef CAS;
(c) P. J. Heard, Prog. Inorg. Chem., 2005, 53, 268 Search PubMed.
- J. Cookson and P. D. Beer, Dalton Trans., 2007, 1459–1472 RSC.
- Y. S. Tan, A. L. Sudlow, K. C. Molloy, Y. Morishima, K. Fujisawa, W. J. Jackson, W. Henderson, S. N. B. A. Halim, S. W. Ng and E. R. T. Tiekink, Cryst. Growth Des., 2013, 13, 3046–3056 CAS.
- P. Cassoux, L. Valade, D. W. Bruce and D. O'Hare, Inorganic Materials, John Wiley & Sons, Chichester U.K., 2nd edn, 1996 Search PubMed.
- A. T. Coomber, D. Beljonne, R. H. Friend, J. L. Bredas, A. Charlton, N. Robertson, A. E. Undrhill, M. Kurmoo and P. Day, Nature, 1996, 380, 144–146 CrossRef CAS.
- D. Fan, M. Afzaal, M. A. Mallik, C. Q. Nguyen, P. O'Brien and P. J. Thomas, Coord. Chem. Rev., 2007, 251, 1878–1888 CrossRef CAS.
- Y. Zhao, W. Perez-Segarra, Q. Shi and A. Wei, J. Am. Chem. Soc., 2005, 127, 7328–7329 CrossRef CAS PubMed.
- G. M. de Lima, D. C. Menezes, C. A. Cavalcanti, J. A. F. dos Santos, I. P. Ferreira, E. B. Paniago, J. L. Wardell, S. M. S. V. Wardell, K. Krambrock, I. C. Mendes and H. Beraldo, J. Mol. Struct., 2011, 988, 1–8 CrossRef CAS.
- J.-G. Kang, J.-S. Shin, D.-H. Cho, Y.-K. Jeong, C. Park, S.-F. Soh, C. S. Lai and E. R. T. Tiekink, Cryst. Growth Des., 2010, 10, 1247–1256 CAS.
- M. K. Bharty, R. K. Dani, P. Nath, A. Bharti, N. K. Singh, O. Prakash, R. K. Singh and R. J. Butcher, Polyhedron, 2015, 98, 84–95 CrossRef CAS.
- X. M. Qian and S. M. Nie, Chem. Soc. Rev., 2008, 37, 912–920 RSC.
- P. Bharati, A. Bharti, P. Nath, M. K. Bharty, R. J. Butcher and N. K. Singh, Polyhedron, 2015, 102, 375–385 CrossRef CAS.
- S. K. Ujjain, P. Ahuja, R. K. Sharma and G. Singh, Int. J. Chem., 2015, 7, 69–82 CAS.
- T. W. Clarkson and L. Magos, Crit. Rev. Toxicol., 2006, 36, 609 CrossRef CAS PubMed.
- R. E. Hoffmeyer, S. P. Singh, C. J. Donan, A. R. S. Ross, R. J. Hughes, I. P. Pickering and G. N. George, Chem. Res. Toxicol., 2006, 19, 753–759 CrossRef CAS PubMed.
-
(a) J. P. K. Rooney, Toxicology, 2007, 234, 145–156 CrossRef CAS PubMed;
(b) J. P. K. Rooney, Toxicology, 2007, 238, 216 CrossRef CAS.
- A. N. Gupta, V. Kumar, V. Singh, A. Rajput, L. B. Prasad, M. G. B. Drew and N. Singh, J. Organomet. Chem., 2015, 787, 65–72 CrossRef CAS.
- N. J. Bulleid and L. Ellgaard, Trends Biochem. Sci., 2011, 36, 485 CrossRef CAS PubMed.
- R. K. Dani, M. K. Bharty, S. K. Kushawaha, S. Paswan, O. Prakash, R. K. Singh and N. K. Singh, J. Mol. Struct., 2013, 1054–1055, 251–261 CrossRef CAS.
- R. K. Dani, M. K. Bharty, S. Paswan, S. Singh and N. K. Singh, Inorg. Chim. Acta, 2014, 421, 519–530 CrossRef CAS.
- C. N. Kapanda, G. G. Muccioli, G. Labar, J. H. Poupaert and D. M. Lambert, J. Med. Chem., 2009, 52, 7310–7314 CrossRef CAS PubMed.
- S. Sharma, R. Bohra and R. C. Mehrotra, J. Crystallogr. Spectrosc. Res., 1991, 21, 61–64 CrossRef CAS.
- B. C. Ranu, A. Saha and S. Banerjee, Eur. J. Org. Chem., 2008, 519–523 CrossRef CAS.
- W. L. Chou, K. H. Yih, G. H. Lee, Y. H. Huang and H. F. Wang, Acta Crystallogr., 2010, 66, o3193 CAS.
- S. Ekthawatchai, S. Kamchonwongpaisan, P. Kongsaeree, B. Tarnchompoo, Y. Thebtaramonth and Y. Yuthavong, J. Med. Chem., 2001, 44, 4688–4695 CrossRef CAS PubMed.
- A. C. Fabretti, G. C. Franchini, C. Preti, G. Toshi and P. Zannini, Transition Met. Chem., 1985, 10, 284 CrossRef CAS.
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier, Amsterdam, 2nd edn, 1984 Search PubMed.
- B. A. Prakasam, K. Ramalingam, G. Bocelli and A. Cantoni, Polyhedron, 2007, 26, 4489–4493 CrossRef CAS.
- N. Srinivasan, S. Thirumaran, S. Kohli and Rajnikant, J. Chem. Crystallogr., 2010, 40, 505–509 CrossRef CAS.
- N. Srinivasan, S. Thirumaran and S. Ciattini, J. Mol. Struct., 2009, 936, 234–238 CrossRef CAS.
- N. Srinivasan, P. J. Rani and S. Thirumaran, J. Coord. Chem., 2009, 62, 1271–1277 CrossRef CAS.
- H. C. M. van Gaal, J. W. Diesveld, F. W. Pijpers and J. G. M. van der Linden, Inorg. Chem., 1979, 18, 3251–3260 CrossRef CAS.
- D. Wang, F. Wang, B. Zhao, Y. Wanga and D. Zhao, RSC Adv., 2015, 5, 89895–89899 RSC.
- C. C. Wu, Y. T. Lin, H. H. Chiang, T. Y. Cho, C. W. Chen, K. T. Wong, Y. L. Liao, G. H. Lee and S. M. Peng, Appl. Phys. Lett., 2002, 81, 577–579 CrossRef CAS.
- A. Bharti, P. Bharati, M. K. Bharty, R. K. Dani, S. Singh and N. K. Singh, Polyhedron, 2013, 54, 131–139 CrossRef CAS.
- A. C. Ekennia, D. C. Onwudiwe and A. A. Osowole, J. Sulfur Chem., 2015, 36, 96–104 CrossRef CAS.
- S. P. Sovil, K. Babic-Samardzija and D. M. Minic, Thermochim. Acta, 2001, 70, 29 CrossRef.
- J. R. Botelho, A. G. Souza, A. D. Gondim, P. F. Athayde-Filho, P. O. Dunstan, C. D. Pinheiro, E. Longo and L. H. Carvalho, J. Therm. Anal. Calorim., 2005, 79, 309–312 CrossRef CAS.
- R. K. Dani, M. K. Bharty, O. Prakash, R. K. Singh, B. Prashanth, S. Singh and N. K. Singh, J. Coord. Chem., 2015, 68, 2666–2681 CrossRef CAS.
- R. K. Dani, M. K. Bharty, S. K. Kushawaha, O. Prakash, R. K. Singh and N. K. Singh, Polyhedron, 2013, 65, 31–41 CrossRef CAS.
- S. H. Dar, S. Thirumaran and S. Selvanayagam, Polyhedron, 2015, 96, 16–24 CrossRef CAS.
- G. A. Ardizzoia, G. L. Monica, A. Maspero, M. Moret and N. Masciocchi, Inorg. Chem., 1997, 36, 2321–2328 CrossRef CAS PubMed.
- M. Nishio, CrystEngComm, 2004, 6, 130–158 RSC.
- G. Hogarth, C. R. Ebony-Jewel, C. R. Rainford-Brent, S. E. Kabir, I. Richards, D. E. T. Wilton-Ely and Q. Zhang, Inorg. Chim. Acta, 2009, 362, 2020–2026 CrossRef CAS.
- P. Bharati, A. Bharti, M. K. Bharty, R. J. Butcher and N. K. Singh, Polyhedron, 2015, 96, 16–24 CrossRef.
- R. Dulare, M. K. Bharty, A. Singh and N. K. Singh, Polyhedron, 2012, 31, 373–378 CrossRef CAS.
- A. Bharti, P. Bharati, R. Dulare, M. K. Bharty, D. K. Singh and N. K. Singh, Polyhedron, 2013, 65, 170–180 CrossRef CAS.
- A. Benedetti, A. C. Fabretti and C. Preti, J. Crystallogr. Spectrosc. Res., 1988, 18, 685–692 CrossRef CAS.
- R. Dulare, M. K. Bharty, S. K. Kushawaha, S. Singh and N. K. Singh, Polyhedron, 2011, 30, 1960–1967 CrossRef CAS.
- S. Gunasekaran, R. A. Balaji, S. Kumaresan, G. Anand and S. Srinivasan, Can. J. Anal. Sci. Spectrosc., 2008, 53, 149–162 CAS.
- B. Kosar and C. Albayrak, Spectrochim. Acta, Part A, 2011, 78, 160–167 CrossRef PubMed.
- M. Belletete, J. F. Morin, M. Leclerc and G. Durocher, J. Phys. Chem. A, 2005, 109, 6953–6959 CrossRef CAS PubMed.
- K. Fukui, Science, 1982, 218, 747–754 CAS.
- D. F. Perepichka and M. R. Bryce, Angew. Chem., Int. Ed., 2005, 44, 5370–5373 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- L. J. Bartolottiand and K. Fluchick, in Reviews in Computational Chemistry, ed. K. B. Lipkowitz and D. Boyd, VCH, New York, 1996, vol. 7, pp. 187–216 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.