
Special catalytic effects of intermediate-water for rapid shock initiation of β-HMX†
Abstract
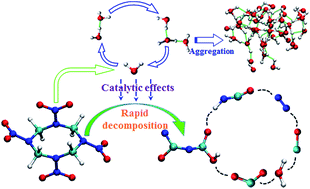
Quantum-based multiscale calculations were carried out to reveal the rapid shock initiation mechanism of β-HMX (octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine). Tracking the dissociation process of HMX, we discovered a special catalytic action of intermediate-water. During early decomposition of HMX, water and its derivative (·OH) acted as oxidizer, dominated the carbon oxidation reaction, and promoted the oxygen-transport from nitrogen to carbon. Water monomer and its small polymers efficiently transferred proton and hydroxyl moieties between reaction centers, also contributing to the acceleration of the carbon oxidation reaction. The carbon oxidation significantly reduced the dissociation energy barrier of C–N bonds, causing fast and deep cracking of HMX. This water catalytic mechanism may contribute to the illustration of the intrinsic difference in reaction properties between sensitive and insensitive explosives, and shed light on understanding the rapid shock initiation process.
 Please wait while we load your content...
Please wait while we load your content...

