
A dynamical coarse-grained model to disclose allosteric control of misfolding β2-microglobulin†
Abstract
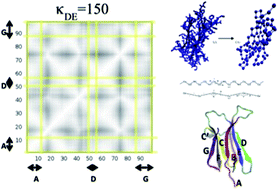
We developed a Coarse-Grained (CG) model for β2-microglobulin using one CG bead per aminoacid residue. The CG model was parameterized using atomistic simulations with the aim of investigating the protein dynamical features related to its natural propensity to misfold and aggregate. The pathological self-aggregation of β2-microglobulin is related to dialysis-related amyloidosis. Despite decades of studies, the mechanism of misfolding and fibrillation of this protein remains controversial. To the best of our knowledge this is the first computational study applying a CG model to follow the structural dynamics of different portions of this amyloidogenic protein in response to external dynamical stimuli. Our results indicate that the propensity to detach the N- and C-terminal β-strands (previously suggested to be part of the fibril formation mechanism) of the protein strongly depend on the strengths of the local interactions between specific internal β-strands. The dynamical changes applied locally to the CG model are able to highlight cooperative rearrangements on selectively coupled regions of the protein, resembling allosteric events, which could provide hints to the understanding of the misfolding mechanism process.
 Please wait while we load your content...
Please wait while we load your content...

