
Behaviour of cation–pi interaction in presence of external electric field†
Nabajit Sarmah and
Pradip Kr. Bhattacharyya*
Department of Chemistry, Arya Vidyapeeth College, Guwahati-781016, Assam, India. E-mail: nsarmah.guchem@gmail.com; prdpbhatta@yahoo.com
First published on 17th October 2016
Abstract
The effect of an external electric field (EEF) on cation–pi interaction between benzene and alkali metal ions has been studied using density functional (DFT) theory and MP2 calculations. Results confirmed that the interaction energy and reactivity of the complexes are sensitive towards the strength as well as direction of the EEF. When EEF is applied perpendicularly to the benzene ring, a linear variation of interaction energy with the field strength is observed. It is further observed that the EEF imparts a significant impact on the curvature of potential energy surface (PES) bringing about a modification on interaction energy. Similarly, the distance of the cation from the pi-ring is also affected by EEF. Results show an inverse dependence of cation–pi distance on the strength of the EEF.
1. Introduction
Noncovalent interactions are abundant in nature and have gained importance in various fields of science viz. chemistry,1–3 biochemistry,4 molecular recognition,5 supramolecular chemistry,6 drug designing,7 protein folding,8 crystal engineering,9 and material science.10,11 Amongst noncovalent interactions, the cation–pi interaction has acquired increasing importance in several fields of contemporary interest, particularly in chemistry, material science and biology.12,13 Several groups11–17 have underscored the significance of cation–pi interactions in diverse fields. However, such interactions were not well recognized until the pioneering work by Doughetry.14–18Cation–pi interaction is essentially electrostatic in origin and strength of this type of interaction is greater than that of other noncovalent interactions.19–21 For example, binding energies for Na+ and Pb+ with benzene are −28.0 and −26.2 kcal mol−1 respectively, which are stronger as compared to the interaction between Na+–H2O (−24.0 kcal mol−1) and Pb+–H2O (−22.4 kcal mol−1).22 Recent studies have also concluded that electron sharing is also instrumental for interaction of an ion with a pi-system. Various other factors like polarity and pH of the solvent, temperature, hydration etc. influence the strength of cation–pi interaction.23–26 Prajapati et al. has measured the contribution of cation–pi interaction on the stability of proteins and based on free energy (ΔG) measurement they concluded that strength of cation–pi interactions increases with increasing temperature.25 Moreover, strength of cation–pi interaction depends on polarity of the solvent; binding energies of the cation–pi complexes of benzene and borazine with alkali metal cations was shown to decrease with increasing solvent polarity.23
It is expected that external perturbations like, radiation, solvents, external electric field (EEF) etc. influence different type interactions. Effect of EEF on chemical reactivity has been, many a times, taken up for study by several research groups.27–35 A recent such study shows that an EEF can influence the interactions containing noncovalent interactions.29 It has also been shown that reactivities of DNA/RNA32 and physical properties of carbon nanomaterials like graphene33 and CNTs34 are also affected by the presence of EEF. Studies also suggest that external perturbation such as an EEF affect anion–pi interactions too.35 The mechanism of ion–pi interaction has recently been studied by Novák et al.36 They advocated that in ion–pi interaction, application of electric field perpendicular to the plane of the pi-system lowers the energy of singlet excited state corresponding to charge transfer between the pi-system and the ion and subsequently increases the covalent nature in the interaction between the two moieties.
Benzene or benzenoid systems are the most common pi-groups which are present in biomolecules and hence studying the effect of an EEF on such systems might be helpful in mimicking such situation particularly in biology. There is a favourable cation–pi interaction for every 77 amino acid residues of protein length and 26% of all tryptophan residues are found to be involved in energetically favourable cation–pi interactions.37
Despite of a rich literatures dealing with cation–pi interactions, studies on the effect of EEF on cation–pi interaction have hitherto been sparse. Therefore, an in-depth study on the effect of EEF on this type of interaction is essential. Herein, we have made an attempt to measure the effect of an EEF on interaction energy, PES and the geometry of the cation–pi complexes using density functional theory (DFT) and MP2 calculations. Benzene is used as a prototypical representation and effect of EEF on benzene–Li+ and benzene–Na+ complexes are studied.
2. Computational details
Geometrical minima of the complexes are obtained in absence as well as in presence of EEF using cc-pVDZ basis set with Becke three parameter exchange and Lee, Yang and Parr correlation functional (B3LYP)38,39 and are confirmed by the absence of any imaginary frequency. A lot of earlier studies have shown the applicability of DFT in describing the cation–pi interactions.40–44 In an article Woon et al. have shown that the basis sets should be augmented with diffuse functions for an accurate description of the molecular properties that affect their noncovalent interactions.45 In view of this, we further optimized the considered system using B3LYP/cc-pVTZ, B3LYP/aug-cc-pVDZ, B3LYP/aug-cc-pVTZ, and B3LYP/6-311+G(2d,2p) level of theories. Studies has shown that MP2 level gives better interaction energy than CCSD although later is much more computationally demanding and recovers more correlation energy.46,47 Therefore we re-optimized the complexes using MP2/cc-pVDZ, MP2/aug-cc-pVTZ, MP2/aug-cc-pVDZ, MP2/aug-cc-pVTZ and MP2/6-311+G(2d,2p) level of theories with the EEF applied perpendicular to the plane of the ring. During the optimization a field strength of 0.03 au has been employed for both benzene–Li+ and benzene–Na+ complexes in +z direction (direction of the axis are shown in Fig. 1). Along with that for benzene–Li+ complex, minima is obtained by applying the field in the −z direction too. Interaction energies are calculated at the chosen level of theories using eqn (1) (mentioned in the Section 3.1). To find out the effect of EEF on PES, PES scan is carried out for benzene–Li+ complex applying an EEF along both +z and −z directions using MP2/cc-pVDZ level of theory. Natural bond orbital (NBO) calculation has also been carried out to calculate the charge transfer during complexation. All the calculations are performed using gaussian09.48 | ||
| Fig. 1 Optimized structure of benzene–Li+ complex (in absence of any external electric field). | ||
3. Results and discussion
The external electric field is applied perpendicular to the plane of the ring (i.e. along z-axis in all the considered complexes). The optimized structure of the cation–pi complex (benzene–Li+ as the representative case) in the absence of any electric field obtained at B3LYP/6-311+G(2d,2p) level of theory along with their Cartesian axes is shown in Fig. 1.3.1 Interaction energy
Recently, it has been confirmed that the super molecular model for calculating interaction energy suffers from serious drawbacks particularly in calculation of interaction energy in presence of EEF. The simple super molecular model has been moderately modified by Foroutan-Nejad et al.,49 who have proposed the corrected equation as in eqn (1).| Einteraction = EFieldcomplex − (EFieldcation + EFieldAr) + FQ(R1 − R0) | (1) |
Interaction energies are calculated using both B3LYP and MP2 levels using the chosen basis set. Obtained interaction energies have been corrected for basis set superposition error (BSSE) using the ‘Counterpoise=N’ as proposed by Boys and Bernardi's counterpoise method.50,51 BSSE corrected interaction energies for benzene–Li+ complex are shown in Table 1 and those for benzene–Na+ complex are in ESI Table S1.† The BSSE correction factors are presented in ESI Table S2.† More negative interaction energy indicates a higher stability of the cation–pi complex towards bond breaking. Values of the interaction energy in absence of any EEF are in good agreement with earlier reported values23 and interaction energy of benzene–Li+ complex is higher as compared to benzene–Na+ complex. However, the interaction energy in benzene–Li+ is less than that found in case of interaction of the Li+ ion with graphene surface.33 With an increase in the field strength along +z direction, interaction energy of the cation–pi complex increases and the reverse is observed when the field is applied along −z direction. Results show a linear variation of interaction energy with the strength of EEF; interaction energy changes by 2 to 3 kcal mol−1 with the change in the field strength by 0.0025 au.
| Field strength (in au) | B3LYP | MP2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| cc-pVDZ | cc-pVTZ | Aug-cc-pVDZ | Aug-cc-pVTZ | 6311+G(2d,2p) | cc-pVDZ | cc-pVTZ | Aug-cc-pVDZ | Aug-cc-pVTZ | 6311+G(2d,2p) | |
| 0.03 | −65.26 | −65.93 | −65.06 | −66.06 | −65.75 | −61.00 | −62.20 | −61.52 | −62.76 | −61.67 |
| 0.0275 | −63.08 | −63.80 | −62.93 | −63.96 | −63.62 | −58.89 | −60.17 | −59.45 | −60.73 | −59.63 |
| 0.025 | −60.88 | −61.66 | −60.75 | −61.81 | −61.47 | −56.76 | −58.13 | −57.35 | −58.65 | −57.56 |
| 0.0225 | −58.68 | −59.50 | −58.54 | −59.64 | −59.29 | −54.62 | −56.07 | −55.23 | −56.54 | −55.49 |
| 0.02 | −56.45 | −57.31 | −56.31 | −57.44 | −57.09 | −11.09 | −53.99 | −53.09 | −54.42 | −53.38 |
| 0.0175 | −54.20 | −55.12 | −54.07 | −55.22 | −54.88 | −13.74 | −51.90 | −50.93 | −52.27 | −51.26 |
| 0.015 | −51.95 | −52.91 | −51.81 | −52.99 | −52.64 | −16.45 | −49.79 | −48.75 | −50.11 | −49.12 |
| 0.0125 | −49.69 | −50.65 | −49.52 | −50.69 | −50.37 | −45.97 | −47.65 | −46.55 | −47.91 | −46.98 |
| 0.01 | −47.41 | −48.38 | −47.21 | −48.39 | −47.84 | −43.78 | −45.49 | −44.31 | −45.69 | −44.75 |
| 0.0075 | −45.10 | −46.07 | −44.85 | −46.04 | −45.62 | −41.56 | −43.30 | −42.03 | −43.42 | −42.51 |
| 0.005 | −42.77 | −43.72 | −42.46 | −43.64 | −43.30 | −39.32 | −41.07 | −39.70 | −41.11 | −40.23 |
| 0.0025 | −40.39 | −41.32 | −40.00 | −41.19 | −40.91 | −37.06 | −38.77 | −37.30 | −38.74 | −37.90 |
| 0 | −37.97 | −38.87 | −37.53 | −38.67 | −38.42 | −34.73 | −36.44 | −34.83 | −36.32 | −35.50 |
| −0.0025 | −35.46 | −36.33 | −34.83 | −36.05 | −35.80 | −32.34 | −34.01 | −32.25 | −33.74 | −33.01 |
| −0.005 | −32.85 | −33.67 | −32.10 | −33.31 | −33.04 | −29.83 | −31.46 | −29.52 | −31.05 | −30.38 |
| −0.0075 | −30.06 | −30.84 | −29.16 | −30.37 | −30.08 | −27.16 | −28.72 | −26.57 | −28.16 | −27.57 |
| −0.01 | −26.98 | −27.74 | −26.31 | −27.28 | −26.80 | −24.22 | −25.68 | −23.23 | −24.91 | −24.41 |
To observe the effect of basis set on interaction energy, we have calculated the interaction energy with five different basis sets using both DFT and MP2 methods. Highest interaction energies (in terms of raw energy) are obtained with cc-pVnZ (where, n = D or T), i.e. basis set having no diffuse function (here raw energy means the energy without BSSE correction). With the introduction of basis set having diffuse function, interaction energy decreases and interestingly lowest interaction energy is shown by aug-cc-pVDZ basis set. The interaction energy follows the order: cc-pVDZ > cc-pVTZ > aug-cc-pVTZ > 6-311+G(2d,2p) > aug-cc-pVDZ. Incorporation of BSSE correction to the interaction energy with all the chosen level of theories produces almost same values of interaction energy. The calculated BSSE corrections show that it increases with the increasing field strength and BSSE corrections are larger for smaller basis set. As for example, for benzene–Li+ complex, at B3LYP/cc-pVDZ level, BSSE correction ranges from 1.27 to 6.77 kcal mol−1 in contrast to 0.39 to 0.79 kcal mol−1 in B3LYP/6311+G(2d,2p) level of theory, ESI Table S2.† The BSSE corrections are even higher when it is calculated at the MP2 level as compared to that obtained with B3LYP values. The calculated values of raw and BSSE corrected interaction energies may vary upto 13.6% at MP2/cc-pVDZ level of theory. Thus, the results suggest that, while calculating the interaction energy, taking care of the BSSE correction is of utmost necessity specially when calculating with smaller basis set. In presence of an EEF, variation of interaction energies in both B3LYP and MP2 levels follow the same trend, but B3LYP level gives higher values of interaction energies. In absence of an EEF too, for benzene–Li+ complex B3LYP/6-311+G(2d,2p) level gives 7.6% higher value of interaction energy as compared to that obtained with MP2 level. When the field strength is increased upto 0.03 au, the difference comes down to 6.2%. Similarly this difference in interaction energy between B3LYP and MP2 level for benzene–Na+ complex is 11.48% and 6.6% in absence and presence of EEF respectively (with field strength 0.03 au). This decrease in difference between the interaction energies calculated at B3LYP and MP2 level of theory is due to the increase in BSSE correction term. That is, field strength and the variation of difference in interaction energies exhibit an inverse relationship in terms of their respective values.
Effect of an EEF is similar to the work done by a mechanical force and it, in turn, affects the potential energy of the system. As put forwarded by an earlier report, the EEF changes the curvature of the potential energy surfaces (PES) and by definition, magnitude of the interaction energy, thus, deviates from that obtained by using simple super molecular model.35 Therefore, we have examined the effect of the EEF on the PES by scanning the distance between the cation and the pi-system in presence of an EEF at MP2/cc-pVDZ level of theory without imposing any constraint on the geometry. Subsequently, interaction energies are calculated from the PES. The scanned PES for the benzene–Li+ complex with the applied EEF along the z-axis without imposing any constraint on the geometry of the complex is shown in Fig. 2.
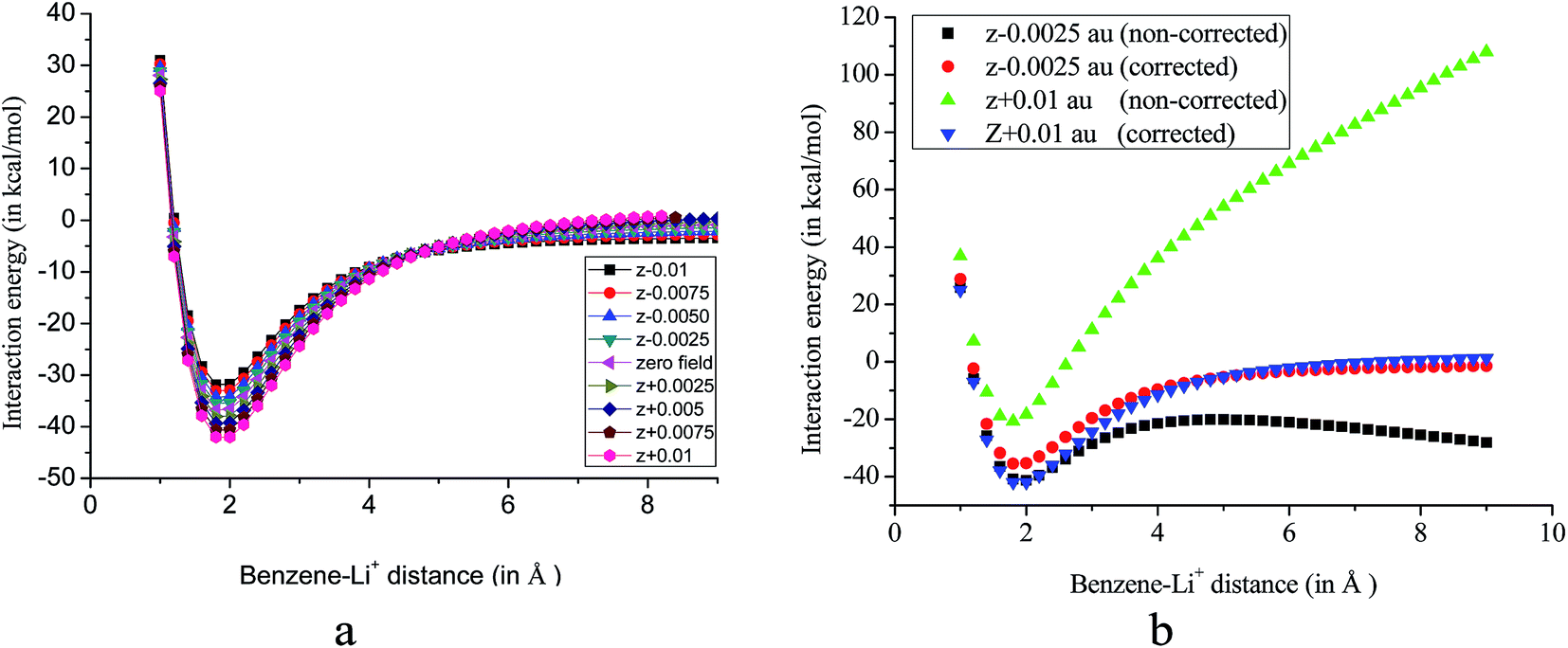 | ||
| Fig. 2 (a) PES (corrected with respect to eqn (1)) for benzene–Li+ complex (b) comparison of corrected and non-corrected PES obtained at MP2/cc-pVDZ level of theory. | ||
PES scan fails to provide correct information regarding the potential energy surface in presence of an EEF unless the electric work required for displacing the charged species at each point on the PES with respect to the origin is considered. This is because in every step of the PES scan, changing the distance of ions with respect to the origin increases the total energy by a factor of QFΔx, where Q and F are the charges and field strength respectively. Δx is a measure of the displacement in presence of an EEF. Foroutan-Nejad et al. asserted that without inclusion of the correction factor, PES can lead to misinterpretation of the result.35 A non-corrected PES (without inclusion of the correction factor) in the presence of an EEF causes a very steep positive or negative curvature near the local minimum and it never reaches a constant potential energy landscape. Corrected set of PES (with the inclusion of the correction factor) is presented in Fig. 2a. The corrected and non-corrected PES collate at the two extreme fields is shown in Fig. 2b. From Fig. 2b, it is further evident that PES of the system behaves in a completely different manner in presence of an EEF and depends on the strength and the direction of the applied EEF.
In addition, we have calculated the interaction energies of benzene–Li+ complex from the PES by applying the electric field of different strengths along the z-axis and the results are compared with that obtained by using eqn (1). Interaction energies from PES are calculated by taking the energy difference of the minima and the plateau. Earlier study has revealed that 99% of significant cation–pi interactions occur within a distance of 6.0 Å.37 In the current study we have scanned the PES upto a distance of 9.0 Å (between the ring and the cation), and assumed that beyond this distance there exist no cation–pi interactions. It is rather impracticable to calculate the interaction energy in the +z direction from the non-corrected PES, since, the potential energy progressively increases and never reaches a constant value. On the other hand, when the field strength is applied along the −z direction, initially, the potential energy decreases and then it starts rising thereafter, again decreases continuously. Interestingly, when PES is corrected with respect to eqn (1), a constant potential energy surface is achieved, Fig. 2a. This explains the applicability of eqn (1) in presence of an EEF. It is worth noting that a plateau is not observed even at the distance of 9.0 Å for the high EEF strength when the field is applied along +z direction. Therefore, we have calculated the interaction energy with EEF applied along the −z direction. The interaction energies calculated from eqn (1) are close to that obtained from the corrected PES. For instance, on application of field strength of 0.0025 au along −z direction calculated interaction energies are −30.53 and −32.34 kcal mol−1 from PES and eqn (1) respectively. Interaction energies calculated from PES in presence of EEF of strengths 0.0050 au, 0.0075 au and 0.0100 au along −z direction are −28.53, −25.40 and −22.34 kcal mol−1 respectively.
To ascertain whether the interaction energy of the system has any relation with the charge transfer during complexation, we have calculated the natural charge densities of the system using the NBO analysis. Charge transfer (Δq) in the system is calculated by taking the difference in charges of the bare cation and that in the cation–pi complex. It is important to note that Δq values show a good correlation (R2 = 0.98) with the calculated interaction energies in the benzene–Li+ complex (with the applied electric field along z-axis) Fig. 3a, which is also consistent with benzene–Na+ complex (R2 = 0.98). When the field is applied along the +z direction, Δq increases (Δq values are provided in ESI Table S3.†) and hence there is an increase in covalent character of the system. The higher interaction energy for benzene–Li+ complex over benzene–Na+ complex can be attributed to charge transfer to a larger extent involved in the former.
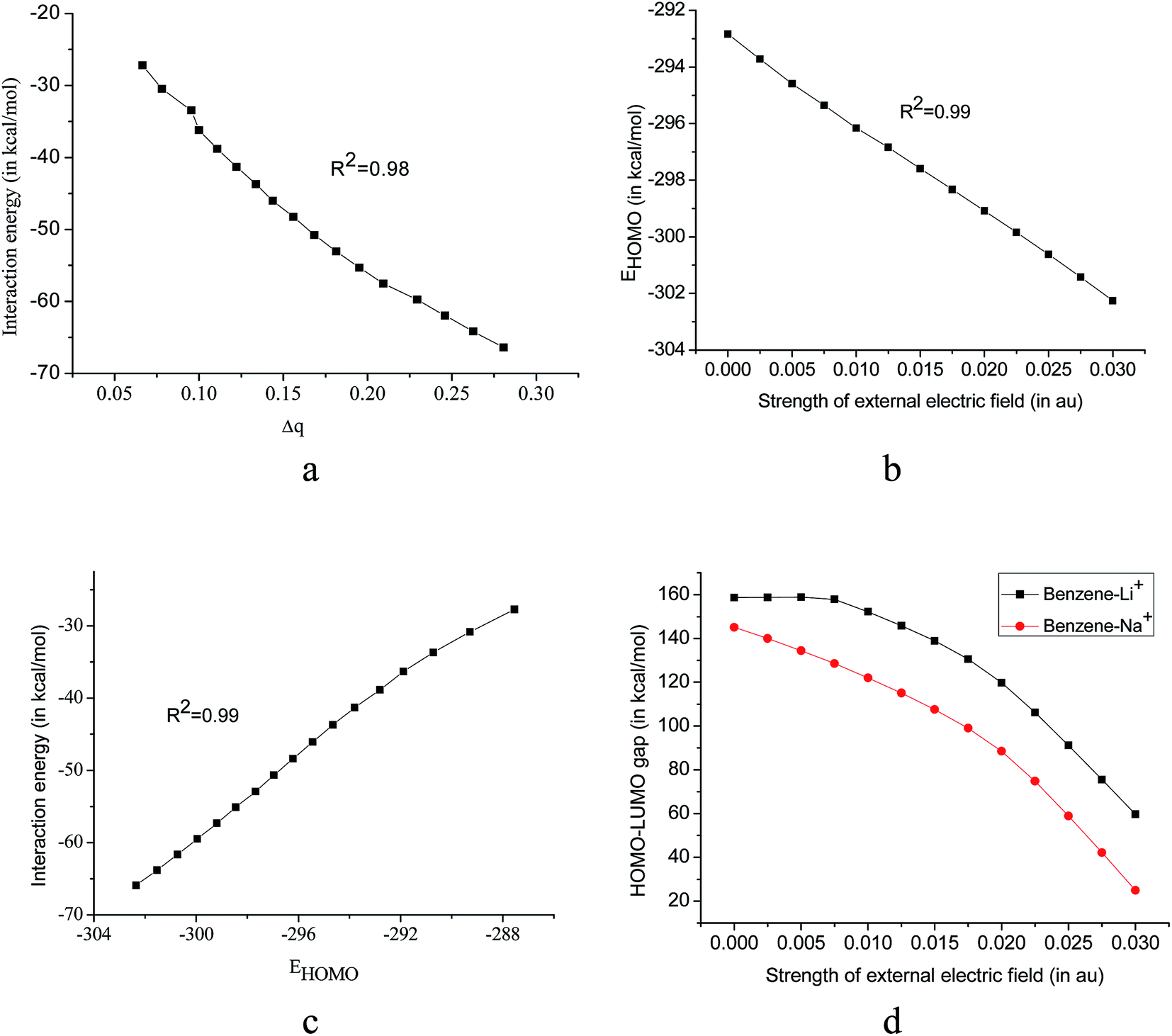 | ||
| Fig. 3 Plot of (a) interaction energy vs. charge transfer (Δq) (b) variation of EHOMO (in kcal mol−1) in presence of EEF (c) interaction energy vs. EHOMO for benzene–Li+ complex (d) variation of HOMO–LUMO gap with in presence of EEF at B3LYP/6311+G(2d,2p) level of theory in presence of EEF along z-axis. | ||
3.2 Variation of HOMO energy of the complexes
Another parameter, which is used to describe the kinetic stability of a system, is HOMO energy (EHOMO) and we observed the variation of EHOMO with the application of the EEF accordingly, the results are presented in Fig. 3b. EHOMO of benzene–Li+ varies linearly with the applied field strength although EHOMO of the benzene–Na+ complex remains almost unaffected. With the increase in the field strength, EHOMO of benzene–Li+ complex decreases i.e. HOMO gets stabilized which is consistent with calculated interaction energies. Interaction energy and EHOMO shows a linear relationship with R2 = 0.99, Fig. 3c.A large HOMO–LUMO gap is indicative of high kinetic stability and low chemical reactivity. Pearson pointed out that the HOMO–LUMO gap represents the chemical hardness of a molecule.52 In the present study, both the systems follow a similar trend in the variation of the HOMO–LUMO gap with EEF. Although EHOMO of the both the system follows different trends, variation in HOMO–LUMO gap is similar. This is because of the compensatory contribution of ELUMO towards constancy in HOMO–LUMO gap.
Application of the field from the opposite direction of the cation (−z direction) does not change the HOMO–LUMO gap much. On the contrary, in presence of field of high strength (beyond 0.01 au in case of benzene–Li+ complex) along +z direction, HOMO–LUMO gap decreases sharply, Fig. 3d. The result is well correlated to the charge transfer in the presence of EEF, i.e. as the strength of EEF increases, HOMO–LUMO gap decreases and charge transfer becomes easier. In the considered systems, the HOMO is largely concentrated on the pi-system and on the other hand LUMO lies over the cation, Fig. 4. Distributions of HOMO and LUMO indicate that the charge-transfer takes place from HOMO to the LUMO.
 | ||
| Fig. 4 Plot of HOMO and LUMO of the benzene–Li+ complex obtained at B3LYP/6311+G(2d,2p) level of theory. | ||
3.3 Thermochemical analysis
To examine the effect of EEF on the thermodynamic driving force, we have carried out thermochemical analysis involved in the complexation. The enthalpy (ΔHcomp) and the Gibbs free energy (ΔGcomp) of the complexation are calculated for all the complexes in presence of EEF (also in absence of any field). ΔHcomp and ΔGcomp are also corrected with respect to eqn (1) and data are presented in Table 2. The value of ΔHcomp in the absence of EEF corroborates well with earlier literature.23 Results show that ΔHcomp is sensitive towards strength and direction of the applied field. On increasing the field strength along +z direction, ΔHcomp value goes on increasing and complexation become more exothermic. However, a complete reversal is observed when the field is applied from the opposite direction (−z direction). For example, upon application of a field of strength 0.01 au along the −z direction of the benzene–Li+ complex (calculated at B3LYP/6311+G(2d,2p) level) ΔHcomp decreases by 11 kcal mol−1 while an increase by 9 kcal mol−1 is observed in presence of EEF of same strength but applied along +z direction. Benzene–Na+ complex exhibits the similar variation and obtained data are provided in ESI Table S4.† It can be inferred from these results that the direction and strength of the applied field affect the thermochemistry of complexation to a significant level.| Field strength (in au) | ΔHcomp | ΔGcomp |
|---|---|---|
| −0.01 | −27 | −19 |
| −0.0075 | −30 | −22 |
| −0.005 | −33 | −25 |
| −0.0025 | −35 | −27 |
| 0 | −38 | −30 |
| 0.0025 | −40 | −32 |
| 0.005 | −43 | −34 |
| 0.0075 | −45 | −37 |
| 0.01 | −47 | −39 |
| 0.0125 | −50 | −41 |
| 0.015 | −52 | −43 |
| 0.0175 | −54 | −46 |
| 0.02 | −56 | −48 |
| 0.0225 | −59 | −50 |
| 0.025 | −61 | −52 |
| 0.0275 | −63 | −54 |
| 0.0300 | −65 | −57 |
The values of thermochemical parameters obtained at B3LYP/cc-pVDZ and B3LYP/cc-pVTZ level of theories show similar variation and are presented in ESI Table S4.† Furthermore, thermochemical data show a higher value of ΔHcomp, and ΔGcomp for benzene–Li+ complex over the benzene–Na+ complex, which underpins stronger interaction of Li+ over Na+ toward the pi ring, Fig. 5. Thus the application of a well directed EEF along +z axis contribute to thermodynamic driving force of complex formation.
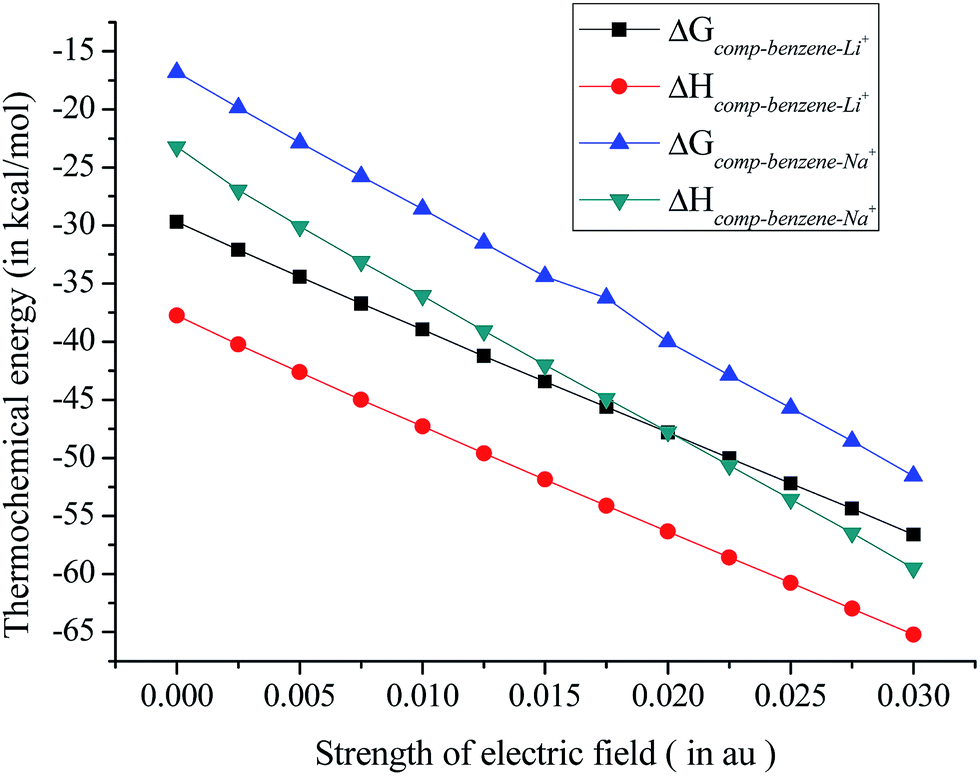 | ||
| Fig. 5 Variation of thermochemical parameters for benzene–Li+ and benzene–Na+ complexes in presence of EEF obtained at B3LYP/6311+G(2d,2p) level of theory. | ||
3.4 Effect of EEF on the geometries of the cation–pi complexes
Several earlier studies have illustrated that EEF can influence the geometry of the complexes. Mata et al. has shown the effect of EEF on the equilibrium geometry of the hydrogen bonded dimer of HF⋯HF.53 It is also learnt from the work of Peles-Lemli et al. that the distance between Li+ and graphene surface is sensitive towards EEF.33 Therefore study of the effect of EEF on the geometries of the cation–pi complexes has also been undertaken. For this purpose geometry of the complexes are re-optimized in presence of EEFs by varying the strength of the field. Since the cation–pi interaction is noncovalent in nature, the cation–pi distance may depend on the level of theory been employed. Accordingly, we have optimized the considered system in five different basis sets using both DFT and MP2 methods. Geometrical parameters for benzene–Li+ complex are presented in Table 3 and for benzene–Na+ complex; values are presented in ESI Table S5.† It is observed that electric field has no considerable effect on the C–C bond length (dC–C) (Fig. 1), but, the distance of the cation from the ring (dM–X) (Fig. 1) is affected to a significant level upon application of EEF. In both DFT and MP2 levels, for benzene–Li+ complex, the dM–X distance follow the order Aug-cc-pVDZ > 6311+G(2d,2p) > Aug-cc-pVTZ. The dM–X distances obtained from the later two levels are almost close to each other and interestingly DFT method gives shorter bond distance as compared to the corresponding values at MP2 level of calculations.| Field strength (in au) | B3LYP | MP2 | ||||
|---|---|---|---|---|---|---|
| Aug-cc-pVDZ | Aug-cc-pVTZ | 6311+G(2d,2p) | Aug-cc-pVDZ | Aug-cc-pVTZ | 6311+G(2d,2p) | |
| 0.03 | 1.6459 | 1.6151 | 1.6170 | 1.6857 | 1.6358 | 1.6415 |
| 0.0275 | 1.6579 | 1.6267 | 1.6285 | 1.6990 | 1.6483 | 1.6540 |
| 0.025 | 1.6712 | 1.6394 | 1.6411 | 1.7132 | 1.6618 | 1.6675 |
| 0.0225 | 1.6859 | 1.6530 | 1.6546 | 1.7284 | 1.6762 | 1.6823 |
| 0.02 | 1.7010 | 1.6675 | 1.6690 | 1.7450 | 1.6918 | 1.6979 |
| 0.0175 | 1.7174 | 1.6831 | 1.6846 | 1.7628 | 1.7085 | 1.7145 |
| 0.015 | 1.7352 | 1.7008 | 1.7016 | 1.7822 | 1.7266 | 1.7326 |
| 0.0125 | 1.7547 | 1.7181 | 1.7201 | 1.8029 | 1.7458 | 1.7521 |
| 0.01 | 1.7758 | 1.7379 | 1.7383 | 1.8260 | 1.7672 | 1.7733 |
| 0.0075 | 1.7989 | 1.7593 | 1.7611 | 1.8506 | 1.7905 | 1.7963 |
| 0.005 | 1.8247 | 1.7829 | 1.7844 | 1.8773 | 1.8162 | 1.8222 |
| 0.0025 | 1.8530 | 1.8094 | 1.8111 | 1.9099 | 1.8433 | 1.8502 |
| 0 | 1.8864 | 1.8423 | 1.8423 | 1.9452 | 1.8776 | 1.8832 |
| −0.0025 | 1.9253 | 1.8732 | 1.8755 | 1.9859 | 1.9152 | 1.9219 |
| −0.005 | 1.9703 | 1.9142 | 1.9163 | 2.0345 | 1.9592 | 1.9667 |
| −0.0075 | 2.0249 | 1.9643 | 1.9652 | 2.0940 | 2.0145 | 2.0224 |
| −0.01 | 2.0966 | 2.0275 | 2.0293 | 2.1720 | 2.0867 | 2.0980 |
With the application of the field along +z direction, an inverse relationship between the dM–X and the field strength is observed. Application of field strength of 0.03 au along +z direction decreases the dLi–X distance by 0.22–0.25 Å and the dNa–X distance by 0.40 Å. This is because the cation moves toward the negative direction of the applied field. On the other hand, the dM–X distance increases as the field is applied along the −z direction. Thus the results confirm that a well directed electric field may impart significant impact on the geometry of a cation–pi complex.
Furthermore, the observed trends are in line with the usual bond length (BL)–bond energy (BE) rule when the correction term in eqn (1) is incorporated during calculation of interaction energy in presence of EEF.
4. Conclusion
The present study illustrates the effect of applied external electric field on cation–pi interactions. Results confirm the sensitivity of the cation–pi interaction towards the direction as well as the strength of the applied field. A marked effect on the interaction energy is observed as the field is applied perpendicular to the ring. This study endorses the fact that to calculate the interaction energy in presence of an external electric field, use of simple supermolecular approach is erroneous. Interaction energy has to be corrected for the cations for their respective distances to the coordinate origin in the complexes. To obtain a plateau of potential energy surface in presence of an external electric field, a similar correction is a pre-requisite. Thermochemical analysis of the considered cation–pi complexes shows that the complexation is exothermic and is observed to be dependent on the strength and direction of the applied external electric field. Besides, the presence of an external electric field exerts a strong influence on the cation–pi distance. However, the impact on C–C bond distances in the pi-system is insignificant. In summary, the strength of the cation–pi interaction as well as the reactivity of the cation–pi complexes can be subjected to manipulation by regulating the magnitude and the direction of the applied external electric field.Acknowledgements
Authors sincerely acknowledge the Department of Science and Technology (DST), Government of India, for financial assistance (Grant No. SB/S1/PC-17/2014).References
- S. Grimme, Chem.–Eur. J., 2012, 18, 9955–9964 CrossRef CAS PubMed.
- M. Juríček, N. L. Strutt, J. C. Barnes, A. M. Butterfield, E. J. Dale, K. K. Baldridge, J. F. Stoddart and J. S. Siegel, Nat. Chem., 2014, 6, 222–228 CrossRef PubMed.
- L. Kronik and A. Tkatchenko, Acc. Chem. Res., 2014, 47, 3208–3216 CrossRef CAS PubMed.
- K. E. Riley and P. Hobza, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 3–17 CrossRef CAS.
- P. S. Murthy, J. Chem. Educ., 2006, 83, 1010 CrossRef CAS.
- J. M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89–112 CrossRef.
- B. R. Beno, K.-S. Yeung, M. D. Bartberger, L. D. Pennington and N. A. Meanwell, J. Med. Chem., 2015, 58, 4383–4438 CrossRef CAS PubMed.
- C. B. Anfinsen, Biochem. J., 1972, 128, 737–749 CrossRef CAS PubMed.
- C. B. Aakeröy, N. R. Champness and C. Janiak, CrystEngComm, 2010, 12, 22–43 RSC.
- K. E. Riley, M. Pitoňák, P. Jurecka and P. Hobza, Chem. Rev., 2010, 110, 5023–5063 CrossRef CAS PubMed.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- D. A. Dougherty, Science, 1996, 271, 163 CAS.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2012, 113, 2100–2138 CrossRef PubMed.
- M. J. Rashkin, R. M. Hughes, N. T. Calloway and M. L. Waters, J. Am. Chem. Soc., 2004, 126, 13320–13325 CrossRef CAS PubMed.
- A. S. Reddy, G. M. Sastry and G. N. Sastry, Proteins: Struct., Funct., Bioinf., 2007, 67, 1179–1184 CrossRef CAS PubMed.
- C. D. Tatko and M. L. Waters, Protein Sci., 2003, 12, 2443–2452 CrossRef CAS PubMed.
- J. Sunner, K. Nishizawa and P. Kebarle, J. Phys. Chem., 1981, 85, 1814–1820 CrossRef CAS.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS PubMed.
- A. S. Reddy and G. N. Sastry, J. Phys. Chem. A, 2005, 109, 8893–8903 CrossRef CAS PubMed.
- G. W. Gokel, S. L. De Wall and E. S. Meadows, Eur. J. Org. Chem., 2000, 2000, 2967–2978 CrossRef.
- G. W. Gokel, L. J. Barbour, L. Stephen and E. S. Meadows, Coord. Chem. Rev., 2001, 222, 127–154 CrossRef CAS.
- B. Guo, J. Purnell and A. Castleman, Chem. Phys. Lett., 1990, 168, 155–160 CrossRef CAS.
- K. K. Bania, A. K. Guha, P. K. Bhattacharyya and S. Sinha, Dalton Trans., 2014, 43, 1769–1784 RSC.
- Q.-S. Du, Q.-Y. Wang, L.-Q. Du, D. Chen and R.-B. Huang, Chem. Cent. J., 2013, 7, 1–8 CrossRef PubMed.
- R. S. Prajapati, M. Sirajuddin, V. Durani, S. Sreeramulu and R. Varadarajan, Biochemistry, 2006, 45, 15000–15010 CrossRef CAS PubMed.
- A. A. Rodríguez-Sanz, E. M. Cabaleiro-Lago and J. Rodríguez-Otero, Org. Biomol. Chem., 2014, 12, 2938–2949 Search PubMed.
- P. K. Chattaraj, Chemical reactivity theory: a density functional view, CRC Press, 2009 Search PubMed.
- K. Kinosita and T. Tsong, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 1923–1927 CrossRef CAS.
- A. Mondal, H. Seenivasan, S. Saurav and A. K. Tiwari, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2013, 52, 1056–1060 Search PubMed.
- H. Hirao, H. Chen, M. A. Carvajal, Y. Wang and S. Shaik, J. Am. Chem. Soc., 2008, 130, 3319–3327 CrossRef CAS PubMed.
- R. Parthasarathi, V. Subramanian and P. Chattaraj, Chem. Phys. Lett., 2003, 382, 48–56 CrossRef CAS.
- B. J. Dutta and P. K. Bhattacharyya, J. Phys. Chem. B, 2014, 118, 9573–9582 CrossRef CAS PubMed.
- B. T. Peles-Lemli, D. N. Kánnár, J. C. Nie, H. Li and S. N. Kunsági-Máté, J. Phys. Chem. C, 2013, 117, 21509–21515 CAS.
- C. Kim, B. Kim, S. M. Lee, C. Jo and Y. H. Lee, Appl. Phys. Lett., 2001, 79, 1187–1189 CrossRef CAS.
- C. Foroutan-Nejad and R. Marek, Phys. Chem. Chem. Phys., 2014, 16, 2508–2514 RSC.
- M. Novák, C. Foroutan-Nejad and R. Marek, J. Chem. Theory Comput., 2016, 12, 3788–3795 CrossRef PubMed.
- J. P. Gallivan and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9459–9464 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- R. A. Kumpf and D. A. Dougherty, Science, 1993, 261, 1708–1710 CAS.
- L. Heginbotham, Z. Lu, T. Abramson and R. MacKinnon, Biophys. J., 1994, 66, 1061 CrossRef CAS PubMed.
- P. Lhoták and S. Shinkai, J. Phys. Org. Chem., 1997, 10, 273–285 CrossRef.
- M. M. Torrice, K. S. Bower, H. A. Lester and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 11919–11924 CrossRef CAS PubMed.
- J.-F. Gal, P.-C. Maria, M. Decouzon, O. Mó, M. Yánez and J. L. M. Abboud, J. Am. Chem. Soc., 2003, 125, 10394–10401 CrossRef CAS PubMed.
- D. E. Woon and T. H. Jr Dunning, J. Chem. Phys., 1994, 100, 2975–2988 CrossRef CAS.
- J. Rezác, K. E. Riley and P. Hobza, J. Chem. Theory Comput., 2011, 7, 2427–2438 CrossRef PubMed.
- P. Hobza and J. Řezáč, Chem. Rev., 2016, 116, 5038–5071 CrossRef PubMed.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, B01, (Gaussian Inc., Wallingford CT, 2010), 2009.
- C. Foroutan-Nejad, M. Novák and R. Marek, J. Phys. Chem. C, 2015, 119, 5752–5754 CAS.
- G. Chalasinski and M. M. Szczesniak, Chem. Rev., 1994, 94, 1723–1765 CrossRef CAS.
- A. Galano and J. R. Alvarez-Idaboy, J. Comput. Chem., 2006, 27, 1203–1210 CrossRef CAS PubMed.
- R. G. Pearson, Hard and soft acids and bases, Van Nostrand Reinhold, 1973 Search PubMed.
- I. Mata, E. Molins, I. Alkorta and E. Espinosa, J. Chem. Phys., 2009, 130, 044104 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Variation of interaction energy, BSSE energy, charge transfer, thermochemical data and dNa–X distance are provided. See DOI: 10.1039/c6ra21334k |
| This journal is © The Royal Society of Chemistry 2016 |