
Design, synthesis and biological evaluation of 2-amino-N-(2-aminophenyl)thiazole-5-carboxamide derivatives as novel Bcr-Abl and histone deacetylase dual inhibitors†
Abstract
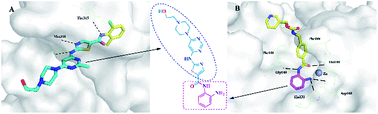
In recent studies, combinations of histone deacetylase (HDAC) inhibitors with kinase inhibitor showed additive and synergistic effects. Herein we present a novel design approach for cancer drug development by combination of breakpoint cluster Abl (Bcr-Abl) and HDAC inhibitory activity, two independent pharmacological activities, in one molecule. The designed compounds were synthesized and tested, showing inhibitory activity against Bcr-Abl and HDAC1. The representative dual Bcr-Abl/HDAC inhibitors, compounds 6a and 6m, showed potent antiproliferative activities against human leukemia cell line K562 and prostate cancer cell line DU145 in cellular assays. This work may lay the foundation for developing dual Bcr-Abl/HDAC inhibitors as potential anticancer therapeutics.
 Please wait while we load your content...
Please wait while we load your content...

