
Effects of temperature, pH and counterions on the stability of peptide amphiphile nanofiber structures†
 *a
Mustafa O.
Guler
*a
and
*a
Mustafa O.
Guler
*a
and
Abstract
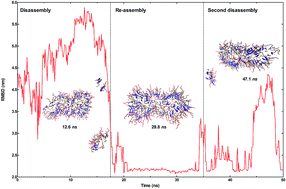
Peptide amphiphiles are a class of self-assembling molecules that are widely used to form bioactive nanostructures for various applications in bionanomedicine. However, peptide molecules can exhibit distinct behaviors under different conditions, suggesting that environmental variables such as temperature, pH, electrolytes and the presence of biological factors may greatly affect the self-assembly process. In this work, we used united-atom molecular dynamics simulations to understand the effects of three counterions (Na+, Ca2+ at pH 7 and Cl− at pH 2) and temperature change on the stability of the lauryl-VVAGERGD peptide amphiphile self-assembly. This molecule contains a bioactive RGD peptide sequence and has been shown to support cellular adhesion and proliferation in vitro. A 19-layered peptide nanostructure, containing 12 peptide amphiphile molecules per layer, was previously shown to exhibit optimal stability and it was used as the model nanofiber system. Peptide backbone stability was studied under increasing temperatures (300–358 K) using the number of hydrogen bonds and root-mean-square deviations of nanofiber size. At higher temperatures, fiber disintegration was observed to be dependent on the type of counter-ion used for nanofiber formation. Interestingly, rapid heating to higher temperatures could sometimes reestablish the integrity of the nanofiber backbone, possibly by allowing the system to bypass an energy barrier and assuming a more thermodynamically stable configuration. As counterion identity was observed to exhibit remarkable effects on the thermal stability of peptide nanofibers, we suggest that these behaviors should be considered while developing new materials for potential applications.
 Please wait while we load your content...
Please wait while we load your content...

