
Probing the electrochemical behavior of {111} and {110} faceted hollow Cu2O microspheres for lithium storage†
Wen-Bei Yu‡
a,
Zhi-Yi Hu‡b,
Ming Yi‡a,
Shao-Zhuan Huanga,
Dai-Song Chena,
Jun Jina,
Yu Li*a,
G. Van Tendelooab and
Bao-Lian Su*acd
aState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, 122 Luoshi Road, 430070, Wuhan, Hubei, China. E-mail: yu.li@whut.edu.cn; baoliansu@whut.edu.cn
bEMAT (Electron Microscopy for Materials Science), University of Antwerp, 171 Groenenborgerlaan, B-2020 Antwerp, Belgium
cLaboratory of Inorganic Materials Chemistry (CMI), University of Namur, 61 rue de Bruxelles, B-5000 Namur, Belgium. E-mail: bao-lian.su@unamur.be
dDepartment of Chemistry and Clare Hall, University of Cambridge, Cambridge CB21 EW, UK. E-mail: bls26@cam.ac.uk
First published on 6th October 2016
Abstract
Transition metal oxides with exposed highly active facets have become of increasing interest as anode materials for lithium ion batteries, because more dangling atoms exposed at the active surface facilitate the reaction between the transition metal oxides and lithium. In this work, we probed the electrochemical behavior of hollow Cu2O microspheres with {111} and {110} active facets on the polyhedron surface as anodes for lithium storage. Compared to commercial Cu2O nanoparticles, hollow Cu2O microspheres with {111} and {110} active facets show a rising specific capacity at 30 cycles which then decreases after 110 cycles during the cycling process. Via advanced electron microscopy characterization, we reveal that this phenomenon can be attributed to the highly active {111} and {110} facets with dangling “Cu” atoms facilitating the conversion reaction of Cu2O and Li, where part of the Cu2O is oxidized to CuO during the charging process. However, as the reaction proceeds, more and more formed Cu nanoparticles cannot be converted to Cu2O or CuO. This leads to a decrease of the specific capacity. We believe that our study here sheds some light on the progress of the electrochemical behavior of transition metal oxides with respect to their increased specific capacity and the subsequent decrease via a conversion reaction mechanism. These results will be helpful to optimize the design of transition metal oxide micro/nanostructures for high performance lithium storage.
1. Introduction
Lithium ion batteries (LIBs) have been widely used in many portable electronic devices, such as mobile phones, laptops and cameras, owing to their high energy and power density, combined with long cycle lifetime. Due to the extremely high reversible capacity and energy density, low cost, and being environmentally-friendly, transition metal oxides have been considered as ideal anodes to replace graphite for lithium ion batteries.1–7Generally, the lithiation and de-lithiation process for many transition metal oxides is a reversible conversion reaction.2,3,7 Therefore, the arrangement of the surface atoms of these transition metal oxide structures is crucial to such a conversion reaction and therefore closely related to their electrochemical performance. From a chemical reaction point of view, the surface structure of electrode materials with exposed high surface energy facets should demonstrate excellent electrochemical performances for LIBs, because more dangling atoms exposed at the surface will facilitate the reaction between transition metal oxides and lithium. For instance, Zhao and co-workers reported that special Fe2O3 octahedra with highly active {111} facets exhibit an increased capacity and excellent high rate cycling performance because of the active {111} facets ensuring electrons and Li+ effectively transferring between the electrolyte and the interface of the Fe2O3 octahedra.8 Sun and co-workers studied nanosized Co3O4 octahedra with enclosed {111} facets for lithium storage and found the capacity slightly increasing during the charge/discharge process, owing to the nanosize and the {111} facets.9 Indeed, this specific capacity increase is widely found for transition metal oxides as anode materials for lithium storage.10,11 And the authors have usually attributed this to the decomposition of the solid electrolyte interface (SEI) and/or the reversible growth of polymeric gel-like layers resulting from kinetically activated electrolyte degradation.9,12 However, from the conversion reaction itself, this explanation is not convincing enough. Recently, our group has synthesized Mn3O4 nano-octahedra, which show not only anomalous magnetic properties and superior photo decomposition activity,13 but also excellent cycling performance and rate capability for LIBs. We proposed that the exposed {011} facets with more dangling Mn atoms facilitate more and more Mn2+ to be converted to Mn3+ during the discharge–charge process.14 This has been further evidenced on the unique walnut-shaped porous MnO2/C nanospheres, which demonstrate a gradual increase of the specific capacity, owing to a deeper conversion reaction for further oxidation from Mn2+ to Mn3+ during the anodic process.15 Therefore, we argue that the capacity increase is mainly from the conversion reaction itself, namely, the enhanced reaction kinetics of Mn2+ to Mn3+ instead of coming from the SEI layer and/or the reversible growth of polymeric gel-like layers.
However, research on the role of the active facets in the electrochemical performance is still seldom reported. To verify our assumption, other transition metal oxides with active facets should be studied. For instance, Xue and co-workers found that the cubic Cu2O with more {100} facets shows the highest capacity and the additional capacity of Cu2O is due to the active facets made it easily oxidized to CuO at the high voltage with the presence of an organic electrolyte.16 However, they only provided CV curves to verify their point. More work should be carried out to probe the electrochemical behaviour of Cu2O with such special active facets for lithium storage.
In this work, we studied the hollow microspheres of Cu2O (HMs-Cu2O) with exposed {111} and {110} active facets as anode materials for LIBs. Such special Cu2O demonstrates an increased capacity during the Li+ insertion/extraction process. Compared to commercial Cu2O nanoparticles without special active facets, HMs-Cu2O delivers not only an enhanced capacity and stability, but also enhanced reaction kinetics with an advanced capacity increase during the Li+ insertion/extraction process. We successfully evidenced that {111} and {110} active faceted Cu2O can be easily oxidized to CuO in the charging process using transmission electron microscope (TEM), high angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) and electron energy loss spectroscope (EELS). However, part of Cu nanoparticles cannot be further converted to Cu2O and CuO during the cycling process. This leads to a capacity decrease in a prolonged cycling process.
2. Experimental section
2.1 Synthesis of hollow Cu2O microspheres
All chemicals are in analytical grade (Aladdin Industrial Corporation, China) and used as received. The reaction is the same as in our previous work.17 For a typical synthesis, 2 mmol of Cu(NO3)2·3H2O (Sinopharm Chemical Reagent Co., Ltd.) is first dissolved in deionized water at a certain volume. Next, 40 mL ethylene glycol (Aladdin Industrial Corporation) is added into the above blue solution. After magnetic stirring for 2 h, the transparent pale blue solution is transferred into 50 mL Teflon-lined stainless steel autoclaves, sealed and transferred to the oven. Then the oven temperature is raised to 180 °C and maintained at that temperature for 1 h. The solid product is filtrated and washed with ethanol, to remove the unreacted ethylene glycol in the final product, and dried at 40 °C in air.For comparison, commercial cuprous oxide nanoparticles (Cu2O-NPs) without specific morphology and active facets were purchased from Aladdin Industrial Corporation (China). From the SEM image shown in Fig. S1,† the particles have a size of ∼50 nm and are tightly aggregated together. HRTEM images shown in Fig. S2† reveal there is no amorphous or other phases in the Cu2O-NPs.
2.2 Materials characterization
Structural and chemical analyses were performed by powder X-ray diffraction (XRD) employing a Bruker D8 Advanced diffractometer with Cu Kα radiation. The morphology of the samples was studied with a Hitachi S-4800 scanning electron microscope (SEM) equipped with a field emission gun (FEG) at an accelerating voltage of 5 kV. Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) were performed on a JEM-2100F transmission electron microscope at 200 kV. High angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) and electron energy loss spectroscope (EELS) were performed on a FEI Titan 80-300 “cubed” microscope fitted with an aberration-corrector for the imaging mode and the probe forming mode, a monochromator, a GIF Quantum energy filter for spectroscopy, operated at 300 kV. The specific surface area is measured using a Tristar II 3020 (Micromeritics, USA) and calculated by the Brunauer–Emmett–Teller (BET) method. The results are shown in Table S1,† which shows the similar low BET surface area of HMs-Cu2O and Cu2O-NPs.2.3 Electrochemical measurements
The anodes are fabricated by mixing the active material, conductive carbon (super P), and polyvinylidene fluoride (PVDF) at a weight ratio of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:10, respectively. N-Methyl pyrrolidone (NMP) is used as a solvent. The resulting slurries are cast onto a copper current collector, and then dried at 120 °C under vacuum for 12 h. The electrode foils are cut into disks with 12 mm in diameter. CR2025 coin-type cells are assembled in an argon-filled glove box by stacking a microporous polypropylene separator containing a liquid electrolyte of LiPF6 (1 M) in ethylene carbonate (EC)/dimethyl carbonate (DMC) (1:1, v/v) between the Cu2O anode and the lithium metal foil cathode. The loading of active material in the composite electrodes is ∼1.0 mg cm−2. The cells are charged and discharged galvanostatically using a battery test system (LAND CT2001A) in a potential range between 0.02 and 3.0 V at various current densities (1C = 400 mA g−1).
:20:10, respectively. N-Methyl pyrrolidone (NMP) is used as a solvent. The resulting slurries are cast onto a copper current collector, and then dried at 120 °C under vacuum for 12 h. The electrode foils are cut into disks with 12 mm in diameter. CR2025 coin-type cells are assembled in an argon-filled glove box by stacking a microporous polypropylene separator containing a liquid electrolyte of LiPF6 (1 M) in ethylene carbonate (EC)/dimethyl carbonate (DMC) (1:1, v/v) between the Cu2O anode and the lithium metal foil cathode. The loading of active material in the composite electrodes is ∼1.0 mg cm−2. The cells are charged and discharged galvanostatically using a battery test system (LAND CT2001A) in a potential range between 0.02 and 3.0 V at various current densities (1C = 400 mA g−1).
3. Results and discussion
Fig. 1 shows the XRD pattern of the as-synthesized product. All peak positions and relative intensities match very well with cubic Cu2O (JCPDS no. 065-3288) and no peak of impurities are found. The sharp narrow peaks also suggest that the products are well crystallized. | ||
| Fig. 1 The XRD result of the as-synthesized HMs-Cu2O. | ||
Fig. 2a presents the typical SEM image of the as-synthesized Cu2O with a uniform diameter of ∼700 nm. Fig. 2b shows one broken microsphere, indicating the hollow structure of the as-synthesized Cu2O. Furthermore, the surface of the Cu2O microspheres clearly reveals that the microspheres are composed of randomly cross-linked polyhedra. Close observation shows that the active facets of {111} and {110} are exposed at the surface. TEM confirms the hollow structure of HMs-Cu2O (Fig. 2c). The lattice fringes observed by HRTEM (Fig. 2d) correspond to 0.245 and 0.302 nm, in agreement with the (111) and (110) planes of Cu2O, respectively. This is consistent with the SEM observations, indicating the exposed highly active {110} and {111} facets at the surface of HMs-Cu2O. It is known that in Cu2O, each oxygen atom is at the center of a tetrahedron of copper atoms, which linearly coordinates with two oxide ions.17,18 Fig. 2e and f demonstrate the surface atomic species and state of {110} and {111} facets respectively, which are composed by either copper cations or oxygen anions.19,20 Our previous study on the absorption of different charged organic chemicals suggests that the {110} and {111} facets are mainly terminated by dangling Cu+ cations for HMs-Cu2O.17 Therefore, the {110} and {111} faceted Cu2O with dangling Cu+ cations is beneficial for the electrochemical reaction of Cu2O: Cu2O + Li → Cu + Li2O. Further, the hollow structure is also helpful for storing the electrolyte and buffering the volume change during the Li+ insertion/extraction process.21,22
 | ||
| Fig. 2 Electron microscopy images of HMs-Cu2O. (a) SEM image at low magnification, (b) one hollow microsphere, (c) TEM image at low magnification and (d) HRTEM image, (e and f) the surface atomic configurations in the {111} and {110} planes of cubic Cu2O. | ||
The electrochemical properties of the hollow Cu2O microspheres are then evaluated. Fig. 3a shows the first four cyclic voltammetry curves of HMs-Cu2O at a scan rate of 0.1 mV s−1 from 0.02 to 3.0 V. At the first scan, a strong cathodic peak at ∼0.9–1.2 V is displayed, corresponding to the chemical reduction reaction of Cu2O → Cu.22 This peak shifts to ∼1.6 V in the following scans, indicating a decreased polarization of the HMs-Cu2O electrode and suggesting an easy electrochemical reverse reaction during the Li+ insertion–extraction process. The weak peak located at ∼0.6 V and that further shifts to ∼0.8 V is attributed to the deep lithiation23,24 and the formation of an SEI layer between Cu2O and the electrolyte.14 At the anodic scan, there are two apparent peaks located at ∼1.4 and ∼2.4 V, which are ascribed to the partial decomposition of SEI and the recombination of Cu and Li2O, respectively.1 These results are in agreement with the discharge–charge profiles at 0.2C (Fig. 3b). In the cycling discharge–charge profiles, we note that with the cycle number increasing, a new charge plateau appears at ∼2.8 V. This plateau may correspond to the further redox reaction between CuO and Cu2O according to previous work25 because the typical phase transformation between Cu2O and Li can be expressed as:
| Cu2O + 2Li+ + 2e− → Li2O + 2Cu | (1) |
| Li2O + 2Cu → Cu2O + CuO + xLi+ + xe− + unreacted Cu | (2) |
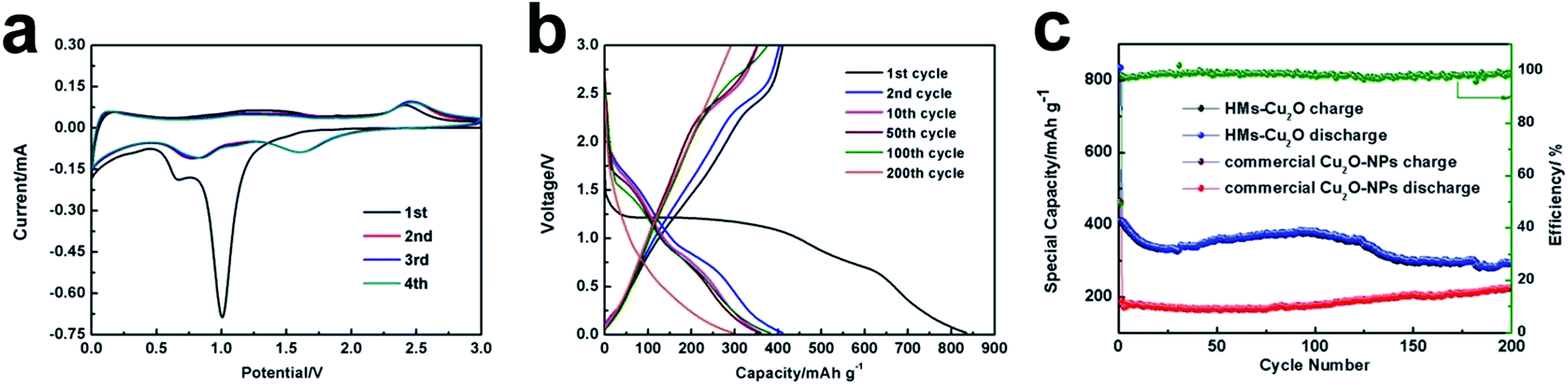 | ||
| Fig. 3 (a) CV response of the HMs-Cu2O electrode at 0.1 mV s−1 for the first four cycles. (b) Discharge–charge voltage profiles for the 1st, 2nd, 10th, 50th, 100th and 200th cycles of HMs-Cu2O at 0.2C. (c) Cycling performance of the HMs-Cu2O with the coulombic efficiency and commercial Cu2O-NPs electrodes at 0.2C. | ||
Fig. 3c displays the cycling performance of HMs-Cu2O and commercial Cu2O-NPs with the cut-off voltages of 0.02–3.0 V (versus Li/Li+) at a current density of 0.2C (1C = 400 mA h g−1). The initial discharge capacity of HMs-Cu2O is 834.7 mA h g−1 at 0.2C, which is much higher than that of commercial Cu2O-NPs (364 mA h g−1). The coulombic efficiency of HMs-Cu2O is also shown in Fig. 3c. It clearly shows the increased coulombic efficiency from the first cycle of 50% to almost 100% after the second cycle. It is interesting to note that the specific capacity slightly increases after 30 cycles for HMs-Cu2O. This is quite different from the previous Cu2O polyhedra with active facets, where no capacity increase is shown.16 After further cycling, the specific capacity gradually decreases from 110 cycles on. Comparing to HMs-Cu2O, the capacity of commercial Cu2O-NPs only exhibits very little increase after 80 cycles. Generally, smaller nanoparticles are beneficial for an enhanced capacity. However, in this experiment, the smaller nanoparticles from commercial Cu2O-NPs demonstrate a lower capacity than that of HMs-Cu2O in microsize. Most probably this is because of the exposed {111} and {110} facets, which can enhance the conversion reaction kinetics. This is similar to previous works on metal oxides with active facets.26 This enhanced reaction kinetics in HMs-Cu2O makes its capacity higher than that of commercial Cu2O-NPs. And the capacity increase largely advances that of commercial Cu2O-NPs. According to the previous work on Cu2O polyhedral with active facets for LIBs16 and formula (2), we suppose that part of Cu2O converts to CuO because of the high theoretical capacity of CuO (674 mA h g−1). This further oxidation explains the capacity increase during the cycling process.
To verify the above speculation, we performed SEM and TEM analysis to follow the structural evolution of HMs-Cu2O during the cycling process. We first selected HMs-Cu2O after 50 cycles at 0.2C because of the improvement of capacity as shown in Fig. 3c. Fig. 4a shows the SEM image of HMs-Cu2O after 50 cycles from the charged state at 0.2C. It clearly displays the well maintained shape and the exposed facets. Fig. 4b provides a close observation of the reacted HMs-Cu2O, showing the SEI film on the surface of the Cu2O microspheres. Fig. 4b inset displays the presence of many small nanoparticles at the surface of the polyhedron after the SEI layer is peeled off. Fig. 4c–f show TEM images of the reacted HMs-Cu2O and the SEI layer, respectively. Fig. 4c clearly shows the presence of some nanoparticles at the surface of the polyhedron. The HRTEM image (Fig. 4d) of the area indicated in Fig. 4c and the corresponding FFT (Fig. 4d inset) reveal the existence of Cu nanoparticles, although the interplanar spacing of Cu (111) (dCu(111) = 0.209 nm) is closed to that of CuO (200) (dCuO(200) = 0.212 nm) and Cu2O (200) (dCu2O(200) = 0.213 nm); however the interplanar spacing of Cu (200) (dCu(200) = 0.532 nm) can be easily detected. Meanwhile, the HRTEM image (Fig. 4f) of the SEI layer and the corresponding FFT (Fig. 4f inset) also demonstrate that some Cu nanoparticles are embedded in the SEI layer, suggesting that the generated Cu nanoparticles infiltrate into the SEI layer from the Cu2O microsphere and cannot be fully converted to Cu2O during the electric cycles. However, the composition and distribution of CuO and Cu2O is still unclear from HRTEM or FFT, because they have very closed lattice spacings (CuO: cubic, a = 4.25 Å; Cu2O: cubic, a = 4.27 Å).
 | ||
| Fig. 4 Electron microscopy images of HMs-Cu2O after 50 cycles for the charge state at 0.2C. (a) SEM image and a Cu2O microsphere with SEI layer (inset), (b) a Cu2O microsphere with partially peeled off SEI layer and corresponding exposed Cu2O microsphere core (inset). (c) TEM image of an exposed Cu2O microsphere core and corresponding low magnification TEM image (inset). (d) HRTEM image of the area indicated in (c) and corresponding FFT pattern, (e) TEM image of the surface of SEI layer from a Cu2O microsphere (inset), and (f) HRTEM image of the area indicated in (e) and corresponding FFT pattern. | ||
The Cu2O and CuO can be differentiated through high resolution EELS analysis, due to the differences of Cu L2,3 edge between Cu1+ and Cu2+.27 Furthermore, by using an annular detector to collect the electrons scattered by the sample overlarge angles, image acquisition and the collection of energy loss scattered electrons using a GIF can be done simultaneously. This make HAADF-STEM-EELS ideally suited for the study of Cu2O/CuO/Cu hybrid as imaging can be directly combined with local chemical information. We performed HAADF-STEM-EELS spectrum analysis on reacted HMs-Cu2O after 50 cycles from the charge state at 0.2C. Fig. 5a shows a HAADF-STEM image of a typical HMs-Cu2O microsphere after 50 cycles, with its corresponding EELS spectrum of the Cu L2,3 edge shown in Fig. 5c (blue colour). An EELS spectrum of the same L2,3 edge from pure CuO (black), pure Cu2O (green) and pure Cu (red) are also shown in Fig. 5c as reference. Note that all spectra, shown in Fig. 5c, have been processed after background removal. The energy shift of the first major maxima peak in energy loss near edge structure (ELNES) of CuO relative to Cu2O and Cu is ∼2.3 eV (Fig. 5c), consistent with a previous report.28 The EELS spectrum of Cu-L3 in HMs-Cu2O after 50 cycles (zone1) presents two split peaks indicated by A and B, which also reveals the co-existence of CuO, Cu2O and Cu. Although the maximum of the first ELNES peaks of Cu2O and CuO occur at the same position, the existence of Cu has been verified by the HRTEM and FFT in Fig. 4. In order to understand the distribution of these components (Cu/Cu2O/CuO), the Cu-L3 edges in the HAADF-STEM-EELS data are further fitted using the EELSMODEL program29 and EELS mapping can be obtained for the different components (Fig. 5d–h). The distribution of Cu, Cu2O and CuO is clearly presented, showing more aggregation of CuO than Cu and Cu2O on the surface of the microsphere. Therefore, combining the TEM and HAADF-STEM-EELS results, we confirm that Cu2O, CuO and Cu co-exist in the HMs-Cu2O after 50 cycles. In addition, Cu nanoparticles appear in the SEI layer and more CuO nanoparticles aggregate on the surface of the polyhedron. Although we only characterized the charge state after 50 cycles, we believe that the Cu nanoparticles are unreversed to Cu2O at the beginning of the reaction. The CuO generated on the surface can enhance the capacity of the electric cycles because of its high theoretical capacity.
 | ||
| Fig. 5 (a and b) HAADF-STEM images of HMs-Cu2O after 50 and 80 cycles for the charge state at 0.2C, respectively. (c) Copper L2,3 core-loss EELS spectra acquired from the indicated areas shown in (a) and (b) compared to referencing Cu compounds: CuO (black curve), Cu2O (green curve) and Cu (red curve) and from different areas indicated in (a) zone1 (50 cycles sample) and in (b) zone2 (80 cycles sample), (d–h) and (i–m) EELS maps of HMs-Cu2O after 50 and 80 cycles at 0.2C, respectively. (e and j) Cu2O (green), (f and k) CuO (red), (g and l) Cu (blue), and (h and m) Cu2O/CuO/Cu overlaid color mapping. | ||
Fig. 3c shows that the capacity of HMs-Cu2O is stable after 70 cycles. We have therefore chosen the charge state after 80 cycles at 0.2C for TEM analysis. After 80 cycles, the microspheres maintain their pristine morphology (Fig. 6a). The Cu nanoparticles in the SEI layer can be easily observed in TEM (Fig. 6b). The edge of the microsphere with a thin SEI layer (amorphous surface, indicated by arrows) is presented in Fig. 6c and the corresponding FFT (Fig. 6d) confirms the co-existence of Cu/Cu2O/CuO. In order to investigate the distribution of the Cu nanoparticles, the inverse FFT filtered image of the Cu (200) ring is presented in Fig. 6f and the location of Cu nanoparticles are indicated by circles in Fig. 6f and c. The corresponding inverse FFT filtered image of CuO/Cu2O (110) and (111) (Fig. 6e) and the overlaid color map reveal the distribution of Cu and CuO/Cu2O on the edge of the microsphere. The Cu (111) ring is not used for the inverse FFT filtering image because of the narrow separation between the dCu(111) and dCuO/Cu2O(200). In order to confirm the existence of CuO and investigate the distribution of the Cu/Cu2O/CuO components, EELS analysis is performed similar with the HMs-Cu2O after 80 cycles. The EELS spectrum (Fig. 5c, zone2) and EELS mapping (Fig. 5i–m) demonstrates still more aggregation of CuO on the surface of microsphere. These results reveal that three different valance states of Cu0, Cu+ and Cu2+ can be observed through TEM and EELS at the charging state after 80 cycles. Compared to reacted HMs-Cu2O for 50 cycles, more Cu nanoparticles appear on the surface of the polyhedron after 80 cycles, while more CuO nanoparticles are still located on the surface of the microsphere. The copper on the surface of the active facets may enhance the conductivity while it is harmful to the specific capacity because the existence of copper may prevent the reverse of more Cu2O in the charging process. However, the presence of CuO is beneficial for the specific capacity because of its high theoretical specific capacity (674 mA h g−1). Our results verify our hypothesis that copper can be further oxidized to CuO via a two-step reaction according to formula (2): Cu converts to Cu2O, which is further converted to CuO. This leads to the specific capacity increase during the cycling process. During this process, more and more CuO nanoparticles ensure the capacity increase although some Cu nanoparticles cannot be reversed to Cu2O and/or CuO.
 | ||
| Fig. 6 TEM of HMs-Cu2O in the charged state after 80 cycles at 0.2C. (a) Low TEM image of a typical microsphere, (b) HRTEM image of a Cu nanoparticle in the SEI film. (c) HRTEM image of the area indicated by a red box in (a). (d) Corresponding FFT of the whole area in (c). (e–g) inverse FFT filtered image of CuO/Cu2O (110) and (111) (e, green), Cu (200) (f, red), and overlaid color map (g). | ||
After 110 cycles, the specific capacity obviously decreases (Fig. 3c). TEM characterization on the electrode after 120 cycles at the charge state (Fig. 7a) shows that the microspheres still maintain their original appearance. A closer view of the surface of the microspheres (Fig. 7b) clearly shows that the compact and smooth active facets have transformed into smaller nanoparticles, similar to the ones after 50 and 80 cycles (Fig. 4c and 6a). Fig. 7c presents a HRTEM image of the surface of the microsphere. The corresponding FFT (Fig. 7c inset) shows a strong intensity ring of Cu (200) and a weak intensity of CuO/Cu2O (111), indicating that more Cu nanoparticles occupy the surface of the microsphere. Fig. 7d displays a HRTEM image of a Cu nanoparticle in the SEI film. The size is similar to that of the Cu nanoparticles after 80 cycles (Fig. 6b). In an intensive HRTEM study, we find more and more Cu nanoparticles in the SEI film and at the surface of the polyhedron. This means that more and more Cu nanoparticles cannot be further oxidized to Cu2O and CuO, which naturally leads to the specific capacity decrease due to more and more loss of active material.
 | ||
| Fig. 7 TEM and HRTEM images of the charge state of HMs-Cu2O after 120 cycles at 0.2C. (a) TEM image of the microspheres. (b) TEM image of the active facets. (c) HRTEM image of Cu nanoparticles and the SEI layer. (d) HRTEM image of a Cu nanoparticle. | ||
To show the advantage of the high active facets on the surface of the microspheres, we further studied the cycle performance of HMs-Cu2O at 1C. This capacity increase process should be similar to the process under 0.2C. The capacity increases after 200 cycles. The specific capacity increases from 150 mA h g−1 at 300 cycles to 450 mA h g−1 at 1500 cycles and then remains stable within a certain number of cycles (Fig. 8). The capacity of HMs-Cu2O is largely over the theoretical capacity of Cu2O (375 mA h g−1). Compared with the highest specific capacity at 0.2C, it has a better performance at 1C. A possible reason is that at higher current density, the redox reaction of HMs-Cu2O becomes deeper at 1C due to the exposed {111} and {110} active facets accelerating the conversion reaction. Namely, more and more Cu2O will be oxidized to CuO during the cycling process at 1C because of the exposed {111} and {110} active facets.
 | ||
| Fig. 8 Electrochemical properties of HMs-Cu2O at 1C. (a) Voltage profile for the 1st, 400th, 1000th and 1600th galvanostatic discharge and charge curves; (b) specific capacity as a function of cycle number. | ||
On the basis of the results and analysis above, we can summarize the electrochemical behaviour of HMs-Cu2O as follows (Fig. 9): (i) according to formula (1) and (2), due to the SEI layer formation and part of the formed Cu nanoparticles in the SEI layer are not reversed to Cu2O, the specific capacity decreases very fast at the beginning of the reaction; (ii) with the reaction proceeding, Cu2O can be further oxidized to CuO and some Cu nanoparticles appear at the surface of the polyhedron. Although part of the formed Cu nanoparticles cannot be reversed to Cu2O, the formation of CuO dominates the reaction cycling process. This leads to the specific capacity increase; (iii) the formation of more and more CuO nanoparticles makes the specific capacity reach a maximum value; (iv) after the stable capacity stage, more and more CuO and Cu nanoparticles are formed, and the unreversed Cu to Cu2O and/or CuO dominates the final process, which leads to the specific capacity decrease.
 | ||
| Fig. 9 The schematic electrochemical behaviour process of the HMs-Cu2O. | ||
4. Conclusions
This work describes how hollow microspheres of Cu2O with {110} and {111} active facets can be beneficial for lithium storage. The as-prepared Cu2O electrodes display an enhanced reversible capacity and cycling stability because more dangling Cu atoms are exposed on the {111} and {110} facets and promote the conversion reaction between cuprous oxide and lithium. We systematically studied the morphological and chemical conversion of the Cu2O microspheres upon cycling via SEM, HRTEM and EELS. At the capacity rising stage, there is the formation of SEI layers, Cu nanoparticles in the SEI layer and CuO nanoparticles at the surface of the polyhedron. With the cycling proceeding, more and more Cu2O is converted to CuO, resulting in a capacity maximum. Finally, more and more Cu nanoparticles cannot be reversed to Cu2O and/or CuO, leading to a capacity decrease. This finding on the electrochemical behaviour for Cu2O may be extended to other transition metal oxides with various phases based on a conversion reaction. We believe that our process presented here is helpful for the design of transition metal oxides based anode materials via a conversion reaction.Acknowledgements
B. L. Su acknowledges the Chinese Central Government for an “Expert of the State” position in the Program of the “Thousand Talents”. Y. Li acknowledges Hubei Provincial Department of Education for the “Chutian Scholar” program. This work is supported by National Key Research Program of China (2016YFA0202602), Program for Changjiang Scholars and Innovative Research Team in University (IRT_15R52) and International Science & Technology Cooperation Program of China (2015DFE52870). Z. Y. Hu and G. Van Tendeloo acknowledge support from the EC Framework 7 program ESTEEM2 (Reference 312483). We thank J. L. Xie, X. Q. Liu and T. T. Luo for the general TEM analysis from Research and Test Center of Materials at Wuhan University of Technology.Notes and references
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- M. V. Reddy, G. V. Subba Rao and B. V. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- A. Vu, Y. Qian and A. Stein, Adv. Energy Mater., 2012, 2, 1056–1085 CrossRef CAS.
- Y. G. Guo, J. S. Hu and L. J. Wan, Adv. Mater., 2008, 20, 2878–2887 CrossRef CAS.
- Y. Li, Z. Y. Fu and B. L. Su, Adv. Funct. Mater., 2012, 22, 4634–4667 CrossRef CAS.
- C.-H. Kuo and M. H. Huang, Nano Today, 2010, 5, 106–116 CrossRef CAS.
- W. Wei, Z. Wang, Z. Liu, Y. Liu, L. He, D. Chen, A. Umar, L. Guo and J. Li, J. Power Sources, 2013, 238, 376–387 CrossRef CAS.
- C. Ding, Y. Zeng, R. Li, Y. Zhang and L. Zhao, J. Alloys Compd., 2016, 676, 347–355 CrossRef CAS.
- G. L. Xu, J. T. Li, L. Huang, W. Lin and S. G. Sun, Nano Energy, 2013, 2, 394–402 CrossRef CAS.
- Y. Sun, X. Hu, W. Luo and Y. Huang, J. Mater. Chem., 2012, 22, 19190 RSC.
- Y. Yu, C. H. Chen, J. L. Shui and S. Xie, Angew. Chem., 2005, 117, 7247–7251 CrossRef.
- S. Laruelle, S. Grugeon, P. Poizot, M. Dollé, L. Dupont and J. M. Tarascon, J. Electrochem. Soc., 2002, 149, A627–A634 CrossRef CAS.
- Y. Li, H. Tan, X. Y. Yang, B. Goris, J. Verbeeck, S. Bals, P. Colson, R. Cloots, G. Van Tendeloo and B. L. Su, Small, 2011, 7, 475–483 CrossRef CAS PubMed.
- S. Z. Huang, J. Jin, Y. Cai, Y. Li, H. Y. Tan, H. E. Wang, G. Van Tendeloo and B. L. Su, Nanoscale, 2014, 6, 6819 RSC.
- S. Z. Huang, Y. Cai, J. Jin, Y. Li, H. E. Wang, L. H. Chen, T. Hasan and B. L. Su, J. Mater. Chem. A, 2016, 4, 4264 CAS.
- K. Chen, S. Song and D. Xue, CrystEngComm, 2015, 17, 2110–2117 RSC.
- D. S. Chen, W. B. Yu, Z. Deng, J. Liu, Y. Li, M. Wu, L. H. Chen and B. L. Su, RSC Adv., 2015, 5, 55520 RSC.
- J. Jin, S. Z. Huang, Y. Li, H. Tian, H. E. Wang, Y. Yu, L. H. Chen, T. Hasan and B. L. Su, Nanoscale, 2015, 7, 12979 RSC.
- J. Jin, S. Z. Huang, J. Shu, H. E. Wang, Y. Li, Y. Yu, L. H. Chen, B. J. Wang and B. L. Su, Nano Energy, 2015, 16, 339–349 CrossRef CAS.
- J. Y. Ho and M. H. Huang, J. Phys. Chem. C, 2009, 113, 14159–14164 CAS.
- C. H. Kuo, C. H. Chen and M. H. Huang, Adv. Funct. Mater., 2007, 17, 3773–3780 CrossRef CAS.
- S. Grugeon, S. Laruelle, R. Herrera-Urbina, L. Dupont, P. Poizot and J. M. Tarascon, J. Electrochem. Soc., 2001, 148, A285–A292 CrossRef CAS.
- C. Q. Zhang, J. P. Tu, X. H. Huang, Y. F. Yuan, X. T. Chen and F. Mao, J. Alloys Compd., 2007, 441, 52–56 CrossRef CAS.
- A. Paolella, R. Brescia, M. Prato, M. Povia, S. Marras, L. De Trizio, A. Falqui, L. Manna and C. George, ACS Appl. Mater. Interfaces, 2013, 5, 2745–2751 CAS.
- J. H. Shin, S. H. Park, S. M. Hyun, J. W. Kim, H. M. Park and J. Y. Song, Phys. Chem. Chem. Phys., 2014, 16, 18226 RSC.
- M. C. Kim, S. J. Kim, S. B. Han, D. H. Kwak, E. T. Hwang, D. M. Kim, G. H. Lee, H. S. Choe and K. W. Park, J. Mater. Chem. A, 2015, 3, 23003 CAS.
- R. D. Leapman, L. A. Grunes and P. L. Fejes, Phys. Rev. B: Condens. Matter Mater. Phys., 1982, 26, 614–635 CrossRef CAS.
- P. L. Potapov and D. Schryvers, Ultramicroscopy, 2004, 99, 73–85 CrossRef CAS PubMed.
- J. Verbeeck and S. Van Aert, Ultramicroscopy, 2004, 101, 207–224 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: SEM and HRTEM images, BET surface areas of commercial Cu2O nanoparticles. See DOI: 10.1039/c6ra21026k |
| ‡ The authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |