
Gene delivery using dendrimer/pDNA complexes immobilized in electrospun fibers using the Layer-by-Layer technique
 a
Helena
Tomás
*a
a
Helena
Tomás
*a
Abstract
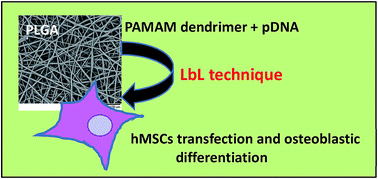
A gene delivery platform for potential use in tissue engineering applications was developed by surface functionalization of biodegradable electrospun poly(lactic-co-glycolic acid) (PLGA) fibers with nanolayers of chitosan (cationic polymer) and alginate (anionic polymer) using the Layer-by-Layer (LbL) technique. The developed system not only supported the attachment and growth of human Mesenchymal Stem Cells (hMSCs), but also was capable of delivering pDNA/dendrimer complexes and inducing cell differentiation towards the osteogenic lineage when a pDNA codifying for human Bone Morphogenetic Protein-2 (BMP-2) was used. Beyond providing a means for pDNA/dendrimer complex immobilization, the polyelectrolyte coating conferred sustained release properties to the scaffold that resulted in pDNA protection from degradation. The polyelectrolyte coating, by itself, also contributed to enhance cell differentiation.
 Please wait while we load your content...
Please wait while we load your content...

