
Three-dimensional porous polyaniline/graphene-coated activated carbon fiber electrodes for supercapacitors†
Hwei-Jay Chu,
Chi-Young Lee and
Nyan-Hwa Tai*
Department of Materials Science and Engineering, National Tsing Hua University, Hsinchu, Taiwan 300, Republic of China. E-mail: nhtai@mx.nthu.edu.tw
First published on 14th November 2016
Abstract
We fabricated unique structured electrodes composed of three-dimensional porous active carbon fibers (ACFs) coated with polyaniline and few-layer graphene (PANI/FLG-coated ACFs), which demonstrated excellent capacity for application in supercapacitors. Ni networks served as the template for graphene grown by annealing the electrodeposited Ni clusters on the surface of ACFs. After chemical vapor deposition, FLG was synthesized on the Ni surface which was subsequently coated with PANI serving as capacitive materials and supporting layers for FLG in the construction of shell structures during Ni etching. The surface combined a pultruded structure and cavity network formed with the PANI/FLG coatings, where the holes on the cavity increased the electroactive surface area and facilitated penetration of the electrolyte into the cavity networks, as a result the capacity was enhanced. In this study, an average specific capacitance of 400.16 F g−1 and an energy density of 61.27 W h kg−1 at a charging current of 0.1 A g−1 over the PANI/FLG-coated ACFs electrodes were achieved.
1. Introduction
Flexible energy-storage devices (ESDs) have attracted much attention recently because of the growing demand for modern consumer electronics requiring the characteristics of light weight, flexibility, and even wearability.1–3 Supercapacitors, also known as electrochemical capacitors, are a promising alternative among various ESDs for energy supply systems, because they exhibit high power capability and good cyclability, and have a short charging time, wide thermal operating range, and low maintenance cost.4–7Supercapacitors are based on two energy-storage mechanisms of electrical-double-layer (EDL) capacitance and pseudocapacitance.6 The capacitance of EDL capacitors arises from the storage of electrostatically separated charges by reversible adsorption of ions at the electrode/electrolyte interface.5 The capacitance of pseudocapacitors originates from fast reversible redox reactions, intercalations, or electrosorption at or near the electrode surface.5,8–10 Therefore, the electroactive surface area (ESA) for ion adsorption is crucial for EDL capacitors.11 Highly porous carbon materials, including graphene and activated carbon,11,12 have extremely high-surface-area structures; besides, single-layer graphene and few-layer graphene (FLG) have drawn enormous interest for their unique electrical, chemical, and mechanical properties.13–16 Among the low-cost electrode materials, activated carbon fibers (ACFs) have been widely used in flexible and textile electrodes for ESDs because of their good mechanical properties and electrochemical performance.17 Although ion transport in EDL capacitors is faster and more reversible than the redox reaction in pseudocapacitors, carbon materials exhibit low energy density because of their limited specific capacitance.6 High energy density can be achieved by using pseudocapacitive materials such as transition-metal oxides and conducting polymers. In contrast to transition-metal oxides, conducting polymers often display higher power density because of their much higher electrical conductivity.18 Polyaniline (PANI) is an interesting conducting polymer that has been extensively used in ESD electrode because of its environmental stability, high electrical conductivity, and ease of synthesis and processing.18–20 Therefore, the electrochemical performance of electrodes used in supercapacitors can be further improved by combining EDL capacitive and pseudocapacitive materials. The graphene/PANI composite electrodes have been widely studied owing to the highly anticipated combination of the two energy-storage mechanisms. The capacitance of graphene/PANI composite electrodes could reach 480 F g−1.20 On the other hand, the PANI/carbon fiber composite electrodes are still attracting for its potential applications in flexible electronics, while they possessed the capacitance of 113 F g−1.23 However, it could be enhanced to over 600 F g−1 of mass-normalized specific capacitance owing to the structural improvement.18
In this study, unique three-dimensional (3D) porous PANI/FLG-coated ACFs were designed, fabricated, and used as electrodes for supercapacitors which with potential applications in flexible and wearable devices. ACFs served as the backbones and the current collectors for flexible electrodes within these systems, and their bare surface could increase the EDL capacitance. Ni networks, prepared by annealing electrodeposited Ni clusters on the ACF surface, were used as templates for graphene grown. After FLG was synthesized on the Ni surface by chemical vapor deposition (CVD), the as-prepared FLG/Ni/ACF composites were coated with PANI layers by dip-coating. PANI layers have faradic contribution and serve as supporting layers for FLG during Ni etching. The shell structures were constructed after Ni was removed from the sites between the ACF surface and FLG layers. The cavity networks formed with PANI/FLG coverings, and the holes on these coverings increased the ESA and facilitated electrolyte penetration into the cavity networks. The performance of the fiber electrodes were significantly improved relative to that of pure ACFs. They also showed better cyclicality and lower self-discharge property, which suggest their potential use as electrodes in flexible ESDs.
2. Experimental section
2.1 Materials
Polyacrylonitrile-based ACFs (part no. AW1114) with diameters of 7–9 μm, conductivity of 10–20 S cm−1, and BET surface of 900–1100 m2 g−1 were purchased from Taiwan Carbon Technology Co., Ltd. All other chemicals used in the study were purchased from Aldrich unless otherwise specified.2.2 Preparation of FLG-coated ACFs
ACFs were refluxed in acetone for three days to remove the polymeric sizing.21,22 After the desizing treatment, the fibers were washed with deionized (DI) water and then dried at 80 °C in an oven overnight followed by Ni coating.The Ni-coated ACFs were fabricated in a stock solution composed of 400 g L−1 NiSO4·6H2O, 45 g L−1 NaCl, and 60 g L−1 H3BO3 by using a potentiostat/galvanostat (Autolab PGSTAT30 & FRA2) operated at −0.9 V for 5, 10, 15, 30 and 45 min at 50 °C. The distance between the ACFs and platinum (Pt) wire counter electrode was fixed at 0.5 cm during Ni electrodeposition. After the Ni-coated ACFs were washed and dried, they were transferred to the CVD system to grow FLG. The samples were heated to 800 °C for 30 min at a heating rate of 50 °C min−1 and a flow of 200 sccm Ar and 300 sccm H2 to anneal the Ni coating. Subsequently, FLG was grown at 1000 °C for 4 min under Ar/H2/CH4 flow (Ar![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2:CH4 = 200:300:5 sccm) at a total pressure of around 25 torr. The system was cooled to 700 °C at around 50 °C min−1 and air-cooled to room temperature in an Ar/H2/CH4 atmosphere.
:H2:CH4 = 200:300:5 sccm) at a total pressure of around 25 torr. The system was cooled to 700 °C at around 50 °C min−1 and air-cooled to room temperature in an Ar/H2/CH4 atmosphere.
The prepared FLG-coated ACFs were used as the reference sample. The electrodeposition time of 30 min for Ni coating was chosen. Coating of the FLG/Ni-coated ACFs with poly(methyl methacrylate) (PMMA) layers was performed by immersing them in a solution of 3.0 wt% PMMA dissolved in acetone. The Ni layer was etched away with 3.0 M hydrochloric acid (HCl) at 90 °C for 3 h. The sample was then washed with DI water and dried in a vacuum pumping system.
2.3 Preparation of 3D porous PANI/FLG-coated ACF electrodes
After FLG on Ni-coated ACFs was synthesized by CVD, they were dip-coated with PANI emeraldine base (PANI-EB) layers at a withdrawal speed of 1 cm s−1. A 0.5 wt% solution of PANI-EB in N-methyl-2-pyrrolidone was used. They were then dried by heating at 80 °C. After four PANI-EB coating cycles, the Ni coating was removed by etching in 3.0 M HCl solution at 90 °C for 5 h, while PANI-EB layers were simultaneously converted into PANI emeraldine salt (PANI-ES) doped with HCl. The sample was washed with DI water and then dried in a vacuum pumping system.2.4 Characterization
The microstructures and morphologies of the electrodes were examined by field emission scanning electron microscopy (FESEM; JSE-6500F and Hitachi-SU8010). The carbon structures of the FLG-coated samples were characterized by using a Raman spectroscope (Horiba HR800 UV) equipped with a 632.8 nm laser source.Electrochemical measurements, namely, cyclic voltammetry (CV), galvanostatic charge/discharge technique, and electrochemical impedance spectroscopy, were performed by using a half-cell system in a three-electrode configuration using Pt wire as counter electrode, Ag/AgCl as reference electrode, and the fiber electrode as working electrode.
3. Results and discussion
The procedure for the preparation of 3D porous PANI/FLG-coated ACF electrodes is schematically shown in Fig. 1. After desizing by acetone (step 1), ACFs were coated with electrodeposited Ni particles (step 2). Before FLG growth in the CVD process (step 3), annealing was performed to facilitate the development of the Ni surface used as template;24 as a consequence, larger Ni grains and Ni coating networks were obtained. Subsequently, FLG was deposited on the annealed Ni-coated ACFs, and PANI-EB layers were then dip-coated onto the surface of the FLG/Ni-coated ACFs (step 4), while PANI covering layers were used as capacitive materials for the additional pseudocapacitance and supporting layers for FLG during Ni etching (step 5). Ni coating networks were removed by etching in HCl solution, while PANI-EB was converted into PANI-ES by HCl doping.25 After the removal of Ni, we found PANI/FLG-coated ACFs that formed cavity networks with the remaining PANI/FLG covering layers. We expected the holes in the layers to facilitate electrolyte diffusion into the cavity and to provide more channels for ion transport.26 | ||
| Fig. 1 Procedure for the preparation of 3D porous PANI/FLG-coated ACF electrodes. | ||
FESEM observations of bare ACFs, Ni-coated ACFs, FLG/Ni-coated ACFs, high magnification of FLG/Ni-coated ACFs, and FLG-coated ACFs are shown in Fig. 2a–e, respectively. Fig. 2a displays the typical morphology of ACFs having shallow grooves on the surface after desizing. Ni clusters containing several particles with sizes ranging from approximately 0.5 to 0.8 μm were observed, and they were arranged on the surface of ACFs during electrodeposition for 30 min (this sample was designated as FLG-coated ACFs-30; Fig. 2b). The Ni clusters led to the formation of Ni networks (Fig. 2c) after annealing. The microstructure evolution of Ni-coated ACFs under different electrodeposition times are shown in Fig. S1.† Ni particles with sizes less than 0.3 μm were distributed sparsely when 5 min of electrodeposition time was adopted. When the electrodeposition time was increased, denser distribution and growth of larger particles was observed. The particles were compactly arranged when ACFs were electrodeposited for 45 min, as a result, formed Ni shell on ACFs after annealing. As shown in Fig. 2c and d, Ni networks was found after annealing the Ni clusters, which served as a substrate for graphene growth. After removal of Ni, the cavity networks were formed with the remaining graphene covering layers (Fig. 2e). Fig. S2† shows FESEM images of the FLG-coated ACFs at various periods of Ni electrodeposition. Isolated cavities with graphene coating were observed in samples with shorter electrodeposition time (see Fig. S2a and S2b†). Moreover, the coaxial core–shell structure of graphene shells was obtained after electrodeposition for 45 min (see Fig. S2c†). While the core–shell structure is used as electrode, worse performance is expected owing to the presence of gaps between ACFs and graphene.
 | ||
| Fig. 2 FESEM images of (a) desized ACFs, (b) Ni-coated ACFs, (c and d) FLG/Ni-coated ACFs, and (e) FLG-coated ACFs-30; (f) Raman spectra of FLG/Ni-coated ACFs. | ||
The carbon structure of the CVD-grown graphene was characterized by Raman spectroscopy (Fig. 2f). After the graphene layers were grown on the Ni-coated ACFs, characteristic graphene fingerprints of D (1325.3 cm−1), G (1572.4 cm−1), D′ (1604.3 cm−1), and 2D (2645 cm−1) bands were observed. The D and D′ bands were caused by structural disorder or edges in graphene, and the G band originated from in-phase vibrations of sp2 carbon atoms in the graphene lattice. The 2D band is the second-order double resonance process related to zone-boundary phonons.27 The level of disorder in graphene may be examined from the intensity ratio of the D band to the G band (ID/IG). The ID/IG ratio of 0.55 in this study and the relatively weak 2D′ band suggest the disordered structure of graphene with vacancy-type defects present in its lattice and reconstructive defects forming at the edges during CVD growth.28 The intensity ratio of the 2D band to the G band (I2D/IG) is an effective indicator for evaluating the number of graphene layers. The I2D/IG ratio of 0.37 we obtained in our study implies the synthesis of FLG with less than four layers.29 As shown in Fig. S3a,† the AFM image indicating that the FLG possesses a thickness of 1.7 nm. In addition, a SEM image with high magnification was supplied for better understanding the FLG morphology.
After dip-coating of PANI layers on the FLG/Ni-coated ACFs by electrodeposition of Ni for 30 min, fibers covered with PANI were obtained (Fig. 3a). According to Fig. 3b–d, the cavity networks formed with the remaining PANI/FLG covering layers where the PANI coatings served as the supporting layers for FLG during formation of shell structures after etching of Ni from the PANI/FLG/Ni-coated ACFs. The TEM image of FLG shows wrinkle microstructure (Fig. S3c†), while FLG covered by PANI depicts that the PANI acted as a supporting layer for FLG (Fig. S3d†). Additionally, the holes on the PANI/FLG covering layers of the shells contribute to the formation of the open pore structure, and the ACFs serve as the current collectors as well as active materials while the covering layers have holes. Therefore, the porous structure possessing a high ESA and high capacitive performance is expected because of facile penetration of the electrolyte into the cavity network.
 | ||
| Fig. 3 FESEM images of (a) FLG/Ni-coated ACFs with PANI coating layers and (b to d) 3D, porous, PANI/FLG-coated ACFs-30. | ||
The electrochemical properties of the ACF-based electrodes and the potential of their use in supercapacitors were investigated by CV and galvanostatic charge–discharge technique, in which a half-cell system using 1.0 M H2SO4 as electrolyte was adopted. Fig. 4a displays a CV comparison of ACFs, PANI-coated ACFs, FLG-coated ACFs-30, and PANI/FLG-coated ACFs-30. The specific capacitance (Cm) can be calculated from the CV curve according to the formula below:30
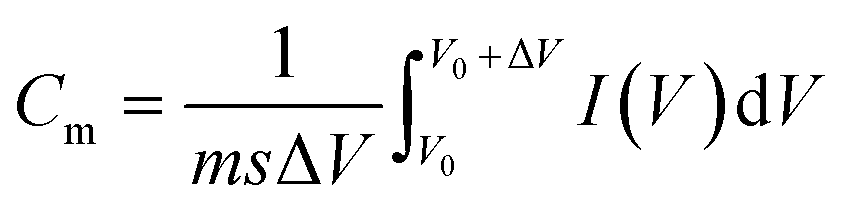 | (1) |
 | ||
| Fig. 4 (a) CV comparison of ACFs, PANI-coated ACFs, FLG-coated ACFs-30, and PANI/FLG-coated ACFs-30 at a scan rate of 5 mV s−1. (b) The calculated specific capacitances of ACFs, FLG-coated ACFs-30, and PANI/FLG-coated ACFs-30 at different scan rates. | ||
The semirectangular CV curve of ACFs showed no obvious redox peaks, indicating the typical electrical EDL behavior of the ACF electrode.6 The quasi-rectangular CV curve of the FLG-coated ACFs-30 electrode implies that the electrochemical behavior of the electrode approaches an ideal supercapacitor and the contribution from pseudocapacitance is negligible, where the FLG film electrode also displayed the quasi-rectangular CV curve (Fig. S4†). The ACFs with PANI-ES coating were prepared to study the supercapacitive performance of ACF-based electrodes. Three pairs of redox peaks can be observed from the CV curve of the PANI-coated ACFs and PANI film electrode (Fig. S4†), which correspond to redox transition of leucoemeraldine/emeraldine forms (O1/R1), hydroquinone/benzoquinone redox reaction of the (O2/R2), and faradic transformation of emeraldine/pernigraniline forms (O3/R3).31 As compared with the Cm of ACFs (136.21 F g−1), PANI film (30.23 F g−1), and FLG film (98.31 F g−1) at a scan rate of 5 mV s−1, those of FLG-coated ACFs-30 (180.34 F g−1) and PANI-coated ACFs (153.25 F g−1) are higher because of the increased surface area and the good electrochemical properties of the coating materials. The semirectangular CV curve of the PANI/FLG-coated ACFs-30 electrode shows a significantly increased CV current, which suggests good capacitance and better charge propagation within the electrode. In addition, the increased area of the closed curves is due to the presence of characteristic redox peaks of PANI; the higher Cm of 243.32 F g−1 may be attributed to faradaic contribution from PANI, the formation of 3D porous structures, and the easy penetration of the electrolyte into the cavity network of the electrode. The calculated Cm of the above samples increased as the scan rate decreased (Fig. 4b).
The ESA can be calculated from the CV curves of electrodes in electrolyte containing 20 mM of K3Fe(CN)6 and 0.2 M of KCl at different scanning rates. The ESA was derived according to the Randle–Sevcik equation:32
| Ip = 2.69 × 105AD1/2n2/3s1/2C | (2) |
 | ||
| Fig. 5 (a) Randles–Sevcik plots of the peak current versus the square root of scan rate for ACFs, FLG-coated ACFs-30, PANI-coated ACFs-30, and PANI/FLG-coated ACFs-30 at different scan rates. The plots show linear relationships between the peak currents and the square root of the scan rates for both electrodes. (b) Ratios of ESA of FLG-coated ACFs-30, PANI-coated ACFs-30, and PANI/FLG-coated ACFs-30 to that ACFs at different scan rates. | ||
The electrochemical performance of the ACF-based electrodes was evaluated galvanostatic charge–discharge tests (Fig. 6). The average galvanostatic specific capacitance (Cave,g) of the electrodes can be obtained from the equation below:33
| Cave,g = IΔt(mΔE)−1 | (3) |
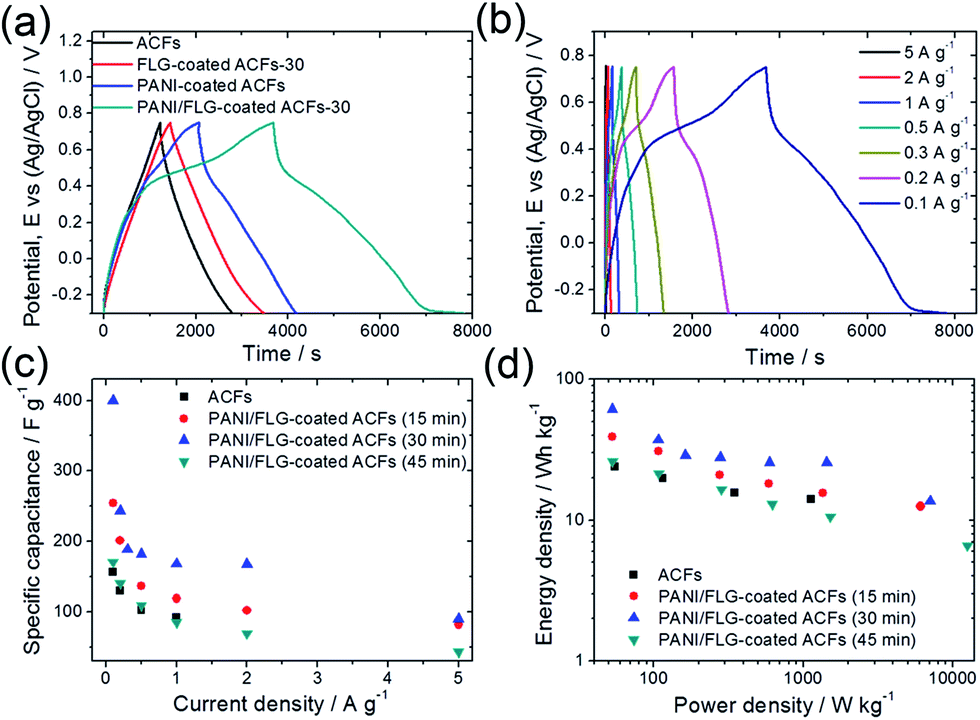 | ||
| Fig. 6 Charge–discharge curves of (a) ACFs, PANI-coated ACFs, FLG-coated ACFs-30, and PANI/FLG-coated ACFs-30 at a charging current of 0.1 A g−1 and (b) PANI/FLG-coated ACFs under various charging currents. (c) Specific capacitances of PANI/FLG-coated ACFs at different Ni electrodeposition times under various charging currents. (d) Comparison of average energy and power densities of ACFs and PANI/FLG-coated ACFs at different Ni electrodeposition times. | ||
The electrochemical performance of PANI/FLG-coated ACFs prepared through different Ni electrodeposition times were studied (Fig. 6c). In comparison with the morphology and microstructure of PANI/FLG-coated ACFs-30 (sample treated for 30 min), the sample treated for 15 min displayed isolated cavities covered with graphene and PANI layers. The sample treated for 45 min exhibited a coaxial core–shell structure of graphene with PANI coating. The electrode treated for 15 min showed higher Cave,g (254.51 F g−1 at 0.1 A g−1) than that of ACFs, which is lower than that of PANI/FLG-coated ACFs-30; while the electrode treated for 45 min performed slightly higher Cave,g (170.27 F g−1 at 0.1 A g−1) than that of ACFs. The results may be explained attributing to the difference microstructures of the electrodes, as shown in Fig. 2, 3 and S2.†
The average energy density (Eave) and power density (Pave) can be derived from the charge–discharge curves according to the following equations:33
| Eave = 0.5C(ΔV)2 | (4) |
| Pave = Eave/Δt | (5) |
The cycling property of PANI/FLG-coated ACFs-30 (Fig. 7a) was investigated under a charging current of 5 A g−1. The capacitance dropped by around 7% after the first 10 cycles, and remained at approximately 92% of capacity retention after 100 cycles. After 1000 cycles, the capacity retention was still kept around 88%, while it remained at about 80% after 2000 cycles. This result may be due to shedding of the PANI/FLG fragments around the holes and irreversible reactions in the first 10 cycles.18 Furthermore, the capacitance dropped by only 5% during the subsequent 1290 cycles (from 11th to 1300th cycle), suggesting good cyclicality of PANI/FLG-coated ACFs-30 after the initial stage. The capacity retention dropped by around 8% from 1300th to 2000th cycle, which could be attributed to the peeling of the PANI/FLG fragments after the high operation cycles.
 | ||
| Fig. 7 (a) Cycling stability of PANI/FLG-coated ACFs-30, (b) Nyquist plots of ACFs and PANI/FLG-coated ACFs-30, and (c) open-potential curve of PANI/FLG-coated ACFs-30 versus time. | ||
In order to evaluate the potential for using PANI/FLG-coated ACFs-30 as ESD electrodes, self-discharge was conducted by charging the electrode to 0.75 V followed by recording the open-circuit potential decay (Fig. 7c). According to the plot, the potential remained at 0.43 V even after 30000 s (∼8.7 h), indicating the low self-discharge property of PANI/FLG-coated ACFs-30.
4. Conclusions
In summary, we prepared 3D porous PANI/FLG-coated ACFs electrodes with a unique structure. After we electrodeposited Ni clusters on the ACF surface, the clusters transformed into Ni networks. These served as a template during annealing. Graphene with few layers was subsequently synthesized on the Ni surface through the CVD method. Before Ni networks were removed from the fibers, they were coated with a PANI layer by dip-coating. These networks behaved as both capacitive materials for the additional pseudocapacitance and supporting layers for graphene during construction of shell structures and Ni etching. The holes found on the PANI/FLG coverings of shells contributed to the formation of the open porous structure. This enhanced the ESA, thereby providing more channels for ion transport and improving capacitive performance by easier penetration of the electrolyte into the cavity network. According to our results, the ESA of 3D porous PANI/FLG-coated ACFs electrodes were twice that of ACFs. The average galvanostatic specific capacitance increased from 156.51 F g−1 (ACFs) to 400.16 F g−1 (PANI/FLG-coated ACFs) at a charging current of 0.1 A g−1. The average energy and power densities were, respectively, 61.27 W h kg−1 and 53.22 W kg−1 at 0.1 A g−1, and 13.7 W h kg−1 and 17.05 kW kg−1 at 5 A g−1. PANI/FLG-coated ACFs showed good cyclicality and low self-discharge property, indicating the potential for using 3D porous PANI/FLG-coated ACFs as electrodes for fabricating flexible ESDs.Acknowledgements
The authors thank the support from the National Science Council, Taiwan, under the contact no. of 104-2221-E-007-029-MY3.Notes and references
- C. Hu, Y. Zhao, H. Cheng, Y. Wang, Z. Dong, C. Jiang, X. Zhai, L. Jiang and L. Qu, Nano Lett., 2012, 12, 5879–5884 CrossRef CAS PubMed.
- J. A. Rogers and Y. Huang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10875–10876 CrossRef CAS PubMed.
- D. Yu, Q. Qian, L. Wei, W. Jiang, K. Goh, J. Wei, J. Zhang and Y. Chen, Chem. Soc. Rev., 2015, 44, 647–662 RSC.
- J. R. Miller and P. Simon, Science, 2008, 321, 651–652 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- J. M. Miller, B. Dunn, T. D. Tran and R. W. Pekala, J. Electrochem. Soc., 1997, 144, L309–L311 CrossRef CAS.
- B. E. Conway, V. Birss and J. Wojtowicz, J. Power Sources, 1997, 66, 1–14 CrossRef CAS.
- T. C. Girija and M. V. Sangaranarayanan, J. Power Sources, 2006, 156, 705–711 CrossRef CAS.
- A. G. Pandolfo and A. F. Hollenkamp, J. Power Sources, 2006, 157, 11–27 CrossRef CAS.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- H. Gwon, H.-S. Kim, K. U. Lee, D.-H. Seo, Y. C. Park, Y.-S. Lee, B. T. Ahn and K. Kang, Energy Environ. Sci., 2011, 4, 1277–1283 CAS.
- T. Ohta, A. Bostwick, T. Seyller, K. Horn and E. Rotenberg, Science, 2006, 313, 951–954 CrossRef CAS PubMed.
- D. Chen, L. Tang and J. Li, Chem. Soc. Rev., 2010, 39, 3157–3180 RSC.
- D. Li and R. B. Kaner, Science, 2008, 320, 1170–1171 CrossRef CAS PubMed.
- P. Liang, L. Yuan, X. Yang, S. Zhou and X. Huang, Water Res., 2013, 47, 2523–2530 CrossRef CAS PubMed.
- Q. Cheng, J. Tang, J. Ma, H. Zhang, N. Shinya and L.-C. Qin, J. Phys. Chem. C, 2011, 115, 23584–23590 CAS.
- Q. Wu, Y. Xu, Z. Yao, A. Liu and G. Shi, ACS Nano, 2010, 4, 1963–1970 CrossRef CAS PubMed.
- K. Zhang, L. L. Zhang, X. S. Zhao and J. Wu, Chem. Mater., 2010, 22, 1392–1401 CrossRef CAS.
- A. Valadez-Gonzalez, J. M. Cervantes-Uc, R. Olayo and P. J. Herrera-Franco, Composites, Part B, 1999, 30, 309–320 CrossRef.
- X.-J. Zhang, W. Su-Qing, T. Yan-Hong, Z. Li and L. Ya-Dong, J. Inorg. Mater., 2013, 28, 836–840 CrossRef.
- D. Lei, K. Devarayan, M.-K. Seo, Y. G. Kim and B.-S. Kim, Mater. Lett., 2015, 154, 173–176 CrossRef CAS.
- D. Q. McNerny, B. Viswanath, D. Copic, F. R. Laye, C. Prohoda, A. C. Brieland-Shoultz, E. S. Polsen, N. T. Dee, V. S. Veerasamy and A. J. Hart, Sci. Rep., 2014, 4, 5049 CAS.
- S.-J. Tang, A.-T. Wang, S.-Y. Lin, K.-Y. Huang, C.-C. Yang, J.-M. Yeh and K.-C. Chiu, Polym. J., 2011, 43, 667–675 CrossRef CAS.
- X. Wang, Y. Zhang, C. Zhi, X. Wang, D. Tang, Y. Xu, Q. Weng, X. Jiang, M. Mitome, D. Golberg and Y. Bando, Nat. Commun., 2013, 4, 2905 Search PubMed.
- D. Graf, F. Molitor, K. Ensslin, C. Stampfer, A. Jungen, C. Hierold and L. Wirtz, Nano Lett., 2007, 7, 238–242 CrossRef CAS PubMed.
- R. K. Biroju and P. K. Giri, J. Phys. Chem. C, 2014, 118, 13833–13843 CAS.
- M. Zheng, K. Takei, B. Hsia, H. Fang, X. Zhang, N. Ferralis, H. Ko, Y.-L. Chueh, Y. Zhang, R. Maboudian and A. Javey, Appl. Phys. Lett., 2010, 96, 063110 CrossRef.
- K. K. Lee, S. Deng, H. M. Fan, S. Mhaisalkar, H. R. Tan, E. S. Tok, K. P. Loh, W. S. Chin and C. H. Sow, Nanoscale, 2012, 4, 2958–2961 RSC.
- S.-B. Yoon, E.-H. Yoon and K.-B. Kim, J. Power Sources, 2011, 196, 10791–10797 CrossRef CAS.
- L. Yuan, M. Yang, F. Qu, G. Shen and R. Yu, Electrochim. Acta, 2008, 53, 3559–3565 CrossRef CAS.
- J. Kim, W.-H. Khoh, B.-H. Wee and J.-D. Hong, RSC Adv., 2015, 5, 9904–9911 RSC.
- B. Abdulhakeem, B. Farshad, M. Damilola, T. Fatemeh, F. Mopeli, D. Julien and M. Ncholu, RSC Adv., 2014, 4, 39066–39072 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra20844d |
| This journal is © The Royal Society of Chemistry 2016 |