
Fluorescent bio-nanocomposites based on chitosan reinforced hemicyanine dye-modified montmorillonite†
Abstract
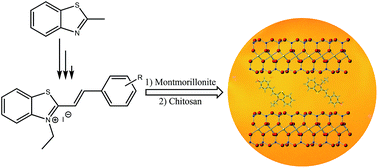
The present investigation describes the synthesis and detailed characterization of novel fluorescent bio-nanocomposite films of chitosan reinforced by hemicyanine dye-modified montmorillonite (MMT–HD) using a solvent-casting method. A series of hemicyanine dyes (HD) have been synthesized by catalyst-free and solvent-free conditions in high yields. These organic compounds were intercalated within the interlayer gallery of montmorillonite providing an increase in the basal spacing and affording inorganic–organic host–guest materials. The fundamental properties of these dyes as well as their intercalated MMT were studied by X-ray, FTIR, and thermal analysis, completed by photophysical properties studies. The results showed that the guest species (HD) are successfully intercalated and stacked as J-aggregates (head-to-tail aggregates) into the host layer of MMT, which is confirmed by spectral characterization. Improvement in thermal stability of the intercalated MMT was indicated by results from thermal analysis compared to the pure dye. Next, the resulting organo-modified clays were dispersed within the chitosan biopolymer. The obtained bio-films were investigated and characterized with varying content of clay (1, 2, 5 and 10 wt%). The structural, hygroscopic (hydrophobicity and water solubility) and fluorescence properties of the resulting materials were evaluated. Results demonstrate that the incorporation of MMT–HD into chitosan improves the hygroscopic properties of the chitosan film, thus showing potential for active food packaging materials. In addition, the solid state fluorescence spectra of the bio-films have shown a significant fluorescence emission at room temperature with increasing clay content.
 Please wait while we load your content...
Please wait while we load your content...

