
Iron piano-stool complexes containing NHC ligands outfitted with pendent arms: synthesis, characterization, and screening for catalytic transfer hydrogenation†
Abstract
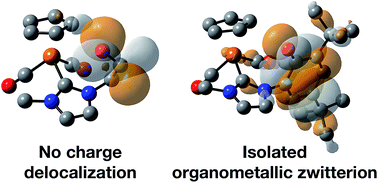
Two iron piano-stool complexes containing NHC ligands equipped with hydroxyl-containing pendent arms (aliphatic and aromatic) have been prepared via deprotonation of the imidazolium salts with a strong base. Access to the anionic ligand is afforded with an additional equivalent of base when the wingtip contains a bulky phenol group. X-ray crystallography and DFT calculations demonstrate an apparent delocalization of the resulting negative charge, which evidently stabilizes the complex. The strongly donating ligands affect the electronic environment at the Fe center to a degree measurable using FTIR spectroscopy. The complexes were screened for catalytic transfer hydrogenation of benzaldehyde, acetophenone and benzophenone using 2-propanol as a solvent and hydrogen donor. The molecular complexes under visible light do not show reactivity towards reduction of carbonyls. Attempts to boost catalyst turnover under in situ conditions using UV light resulted in an unwanted photo-induced pinacolization of acetophenone in both cases and decomposition of the aromatic-containing NHC complex to an iron mirror.
 Please wait while we load your content...
Please wait while we load your content...

