
Temperature-dependent 51V nuclear magnetic resonance spectroscopy for the positive electrolyte of vanadium redox flow batteries
Soohyun Kim,
Chanyong Choi,
Riyul Kim,
Hyun Gyu Kim and
Hee-Tak Kim*
Department of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology, 291 Daehak-ro, Yuseong-gu, Daejeon 305-701, Republic of Korea. E-mail: heetak.kim@kaist.ac.kr
First published on 6th October 2016
Abstract
Temperature-dependent 51V nuclear magnetic resonance (NMR) spectroscopy is used to study the high temperature stability of the VO2+ positive electrolyte of vanadium redox flow batteries (VRFBs). The NMR spectra at high temperatures feature significant line broadening of the VO2+ signal and a narrow line from VO(OH)3. The temperature, acid concentration, and VO2+ concentration dependencies of the line broadening collectively indicate the formation of paramagnetic VO2+ with increasing temperature and consequent paramagnetic dipolar broadening. In order to more clearly monitor the signal from VO(OH)3, which is indicative of the thermal instability of the VO2+ electrolyte, paramagnetic dipolar broadening of the VO2+ signal is intentionally induced by adding an appropriate amount of VO2+. This new analysis shows that, contrary to the previous perception, VO(OH)3 exists even at room temperature. The induced paramagnetic dipolar broadening can be utilized to assess approaches to improve temperature stability of the vanadium electrolyte.
1 Introduction
Owing to its ability to store large amounts of energy and its flexible modular design, the vanadium redox flow battery (VRFB) is one of the most promising types of battery for use in energy storage systems.1 Furthermore, VRFBs have the advantages of long cycle life, short response time, and no cross-contamination.2–4 Currently, VRFB technologies are evolving toward more energy-efficient and reliable systems through improved material development, better stack design, and more suitable operation strategies.5 However, the vanadium concentration of the VRFB electrolyte, which is a decisive factor for the energy density of the VRFB, is limited by vanadium ion solubility and temperature stability. Furthermore, because of the requirement of heat management to control the operation temperature in a narrow range, the temperature stability significantly influences the VRFB cost. In this regard, research on vanadium electrolytes has been directed toward increasing the vanadium concentration and improving the temperature stability. The main limitation to increasing the vanadium concentration is the thermal precipitation of VO2+ (V5+) ions in the positive electrolyte at elevated temperatures. VO2+ ions are rather stable at low temperatures, but become unstable above 40 °C. These solubility characteristics limit the operation temperature to a range of 10–40 °C. For more reliable operation, the vanadium concentration for practical VRFBs is therefore usually not higher than 1.5 M.6 Vijayakumar et al.7 used the density functional theory (DFT) calculations and the temperature dependent chemical shifts in 51V and 17O nuclear magnetic resonance (NMR) spectroscopy to suggest that VO2+ ion in a sulfuric acid supporting electrolyte exists in the form of VO2(H2O)3+, which may undergo a deprotonation reaction and yield a neutral species, VO(OH)3, through an endothermic reaction, as shown in eqn (1). When the temperature increases, VO(OH)3 can be precipitated in the form of solid V2O5 through a condensation process, as shown in eqn (2).
 | (1) |
| 2VO(OH)3 → V2O5·3H2O↓ | (2) |
In order to improve the high temperature stability of the VO2+ electrolyte, various approaches including the use of hydrochloric acid as a supporting electrolyte8,9 and the use of various additives10–13 have been suggested. The improved stability in the presence of HCl was explained by the formation of VO2(OH)2Cl, which can prevent deprotonation.
In these studies on the destabilization mechanism and on electrolyte development toward better stability, 51V NMR spectroscopy has played an important role as an indicator of physical and chemical changes of the vanadium ion. The only oxidation state of vanadium ions active in 51V NMR is VO2+, owing to its diamagnetism. Indeed, the other oxidation states (V2+, V3+, and VO2+) are paramagnetic and give rise to an extreme broadening of the vanadium NMR lines.14 In the interpretation of the NMR spectra, the chemical shift is mainly adopted to monitor any chemical and physical changes. Vijayakumar et al.7 regarded an abrupt change in temperature dependency of the chemical shift as indicative of a transition from VO2(H2O)3+ to neutral VO(OH)3; however, there were no data or comments on the temperature dependency of the NMR signal intensity or the line width. S. Kim et al.8 reported significant line broadening of the 51V NMR signal for an HCl containing electrolyte at high vanadium concentrations and high temperatures; they suggested that the line broadening of the VO2+ electrolyte is due to the production of a small amount of VO2+ caused by exposure to ambient air and consequent paramagnetic dipole broadening. However, experimental evidence for the postulation was not provided.
Against this backdrop, we analysed the 51V NMR spectra of the VO2+ electrolyte with varying temperature, and paid more attention to the line broadening, which had been almost neglected in previous analyses. Our approach is based on the expectation that line broadening originates from paramagnetic dipole broadening due to dynamic exchange of VO2+ and VO2+ and that, by exploiting such line broadening for VO2+, a clearer detection of VO(OH)3 can be achieved. For this purpose, first, we demonstrate significant line broadening of the VO2+ solution in the presence of VO2+, and consequent clearer detection of the peak from VO(OH)3, which is less sensitive to paramagnetic line broadening. Next, to provide evidence for temperature-induced VO2+ formation and consequent paramagnetic dipolar broadening, we describe temperature-induced paramagnetic dipolar broadening for 0.5 M and 2.0 M VO2+ solutions. Finally, we demonstrate that the evolution of VO(OH)3 can be more clearly detected by adding a small amount of VO2+ to the VO2+ solution; this method provides a new methodology to investigate the high temperature stability of the positive vanadium electrolyte.
2 Experimental
A 2.0 M VO2+ electrolyte was prepared by dissolving an equivalent amount of vanadyl sulfate (VOSO4, energy storage) in 4.0 M sulfuric acid solution, which was then electrochemically charged using a VRFB single cell to prepare 2.0 M VO2+/6.0 M total sulfate electrolyte. A 0.5 M VO2+/4.5 M total sulfate electrolyte and a 0.5 M VO2+/1.5 M total sulfate electrolyte were prepared by diluting the 2.0 M VO2+/6.0 M total sulfate electrolyte with 4.0 M sulfuric acid and deionized water, respectively. The 2.0 M VO2+ electrolyte and the 2.0 M VO2+ electrolyte were mixed to make VO2+/VO2+ mixture electrolytes. The electrolyte for monitoring the temperature dependency of VO(OH)3 was prepared by mixing the VO2+ and VO2+ electrolytes at a total vanadium concentration of 2.5 M and at a VO2+/VO2+ ratio of 2.0/0.5. 51V NMR spectra were recorded using a liquid 400 MHz NB NMR spectrometer (Agilent 400 MHz 54 mm NMR DD2) at various temperatures; the chemical shift was externally referenced to VOCl3. Temperature-dependent 51V NMR spectra were collected stepwise with increasing temperature.3 Results and discussion
3.1 51V NMR spectra of the mixture solutions of VO2+ and VO2+
The influence of paramagnetic VO2+ (V4+) on a 51V NMR signal from VO2+ was investigated with the mixtures of VO2+ and VO2+ at various mixing ratios. In these circumstances, exchange between a diamagnetic molecule and a paramagnetic molecule can be described as below:| VO2+ (diamagnetic) + VO2+ (paramagnetic) ↔ VO2+ (paramagnetic) + VO2+ (diamagnetic) | (3) |
The vanadium nucleus experiences only the external magnetic field while in the diamagnetic state. When the vanadium nucleus is in its paramagnetic state, it experiences the external field plus an additional field due to hyperfine electron–nuclei interaction. The rapid exchange of vanadium nuclei between these magnetic states results in a broadening of the NMR line from its diamagnetic state. The general equation that accounts for the influence of the exchange reaction on the line width (1/T2) can be written as below:15–17
 | (4) |
In this expression, tD is the lifetime of the diamagnetic state (VO2+), tP is the lifetime of the paramagnetic state (VO2+), and A is the hyperfine coupling constant of the vanadium nuclei. As the VO2+ concentration increases, tD becomes shorter and tP lengthens, causing line broadening.
The room temperature 51V NMR spectra for the VO2+/VO2+ mixed electrolytes of 2.0 M vanadium and 6.0 M total sulfate concentration and their intensity-normalized spectra are shown in Fig. 1(a) and (b), respectively. The VO2+/VO2+ ratio was varied to have values of 0.0/2.0, 0.1/1.9, 0.2/1.8, 0.5/1.5, and 1.0/1.0, which correspond to states of charge (SOC) of 100%, 95%, 90%, 75%, and 50%, respectively. The VO2+-free electrolyte (2.0 M VO2+/6.0 M total sulfate) exhibited a peak centered at −585 ppm against the VOCl3 reference. With the increase of the VO2+/VO2+ ratio, the intensity of the VO2+ line broadened and, at the ratio of 0.5/1.5, the signal disappeared in spite of the high VO2+ concentrations (Fig. 1(a)). This demonstrates that the line broadening is quite sensitive to the presence of VO2+. There can be no line broadening due to reduced mobility because the viscosity of the electrolyte was not significantly changed with the ratio change. Therefore, the line broadening behaviour results from paramagnetic dipolar broadening of the VO2+ signal in the presence of VO2+.
 | ||
| Fig. 1 51V NMR spectra of 2.0 M vanadium + 6.0 M total sulfate solution with different VO2+/VO2+ ratio: (a) absolute line intensities, (b) normalized line intensities, and (c) line width and chemical shift as a function of the VO2+ concentration in the 2.0 M vanadium electrolytes. | ||
Interestingly, as the line intensity was reduced, a sharp line at −532 ppm appeared more clearly in the intensity-normalized spectra (Fig. 1(b)). The signal is assigned to VO(OH)3 according to the results of previous work.7 M. Vijayakumar et al. used DFT calculation to suggest that the chemical shift of VO(OH)3 is higher than that of VO2+.7 Moreover, VO(OH)3 is well known to exist at room temperature and at low pH (∼3.5).18,19 The line at −532 ppm was weak, indicating the low concentration of VO(OH)3 in the solutions. The narrow line also suggests that the NMR signal of VO(OH)3 is not affected by the paramagnetic dipolar broadening and, in order words, VO(OH)3 is not dynamically exchanged with VO2+.
In the plots of the chemical shift and the line width vs. VO2+ concentration (shown in Fig. 1(c)), an abrupt change in chemical shift can be identified. This change in chemical shift was previously used as an indicator of a physical or chemical change of VO2+.7–9,12 The above results indicate that the degree of paramagnetic broadening can also induce such a transition in the chemical shift and line broadening. Therefore, in the chemical shift analysis of the 51V NMR signal from the vanadium electrolyte, the paramagnetic dipolar broadening effect is carefully taken into consideration.
3.2 Temperature-dependent 51V NMR spectra of the 0.5 M and 2.0 M VO2+ electrolytes
Line broadening for the VO2+ solution at high temperatures has not been explained yet. We suggest that it originates from the generation of VO2+ from VO2+ with increasing temperature. In a VO2+ solution, VO2+ can be formed at low levels of pH and high temperatures via the following reaction:
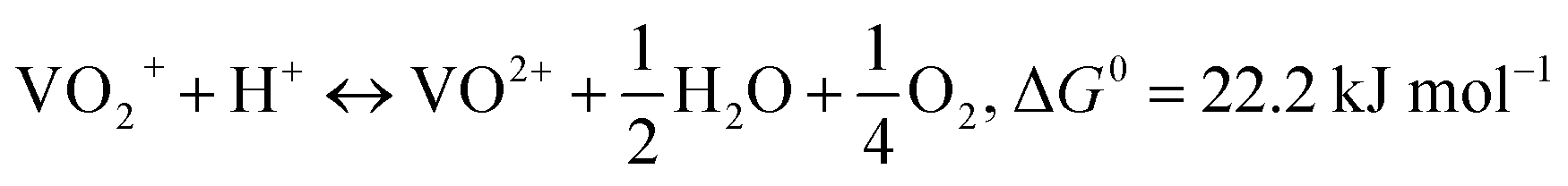 | (5) |
According to the equation, the equilibrium moves toward more VO2+ with increasing temperature and increasing H+ and VO2+ concentrations. The formation of VO2+ and consequent line broadening are evidenced by the dependence of the line broadening on the sulfuric acid concentration. Fig. 2(a) and (b) show the temperature-dependent 51V NMR spectra for the 0.5 M VO2+/4.5 M total sulfate and 0.5 M VO2+/1.5 M total sulfate electrolyte, respectively. The two solutions have the same VO2+ concentration; however, the 1.5 M sulfate electrolyte has a lower H+ concentration than that of the 4.5 M sulfate electrolyte. As can be seen in Fig. 2(a), for the 0.5 M VO2+/4.5 M total sulfate electrolyte, line broadening with temperature increase was clearly observed. In sharp contrast to this, for the 0.5 M VO2+/1.5 M total sulfate electrolyte (Fig. 2(b)), such line broadening did not happen. Rather, the signals became narrower with increased temperature due to increased mobility. The dependence of the H+ concentration on the temperature-dependent 51V NMR signal supports the above VO2+ formation mechanism. For both the 0.5 M VO2+ solutions, no peak from VO(OH)3 appeared (see Fig. 2(a) and (b)), indicating that the deprotonation of VO2+ to form VO(OH)3 was not significant at such a low VO2+ concentration.
 | ||
| Fig. 2 Temperature-dependent 51V NMR spectra of (a) 0.5 M VO2+/4.5 M total sulfate electrolyte, (b) 0.5 M VO2+/1.5 M sulfate electrolyte, and (c) 2.0 M VO2+/6.0 M total sulfate electrolyte. | ||
Fig. 2(c) shows the temperature-dependent 51V NMR spectra for the 2.0 M VO2+/6.0 M total sulfate electrolyte, which is known at high temperature to be more unstable than the 0.5 M VO2+/4.5 M total sulfate electrolyte. As shown in Fig. 2(c), for a highly concentrated VO2+ solution (2.0 M VO2+/6.0 M total sulfate), the broadening in 51V NMR with increasing temperature was more significant than that for the 0.5 M solution with increasing temperature. It can be understood that the forward reaction in eqn (5) is favoured with increasing VO2+ concentration. Contrary to the case of the 0.5 M VO2+ solution, the 2.0 M VO2+ solution exhibited the peak from VO(OH)3; above 303 K, this peak has a lower line width and higher chemical shift (∼−535 ppm) than those from VO2+. With increasing temperature, the peak could be more clearly identified due to the paramagnetic dipolar broadening of the VO2+ peak. However, this peak was hardly seen below 303 K because of overlap with the peak from VO2+, although the presence of VO(OH)3 at low temperatures is highly probable considering the appearance of the peak for the VO2+/VO2+ mixture at room temperature (see Fig. 1(b)).
3.3 Monitoring of VO(OH)3 peak by adding VO2+ into VO2+ electrolyte
The line broadening of VO2+ allows us to explore a way to monitor VO(OH)3 more clearly; by adding VO2+ into the VO2+ electrolyte, line broadening of the VO2+ peak can be intentionally induced, minimizing the overlap of the two lines.For instance, instead of a 100% SOC positive electrolyte (VO2+/VO2+ = 10/0), an 80% SOC positive electrolyte (VO2+/VO2+ = 8/2), which has the same concentration of VO2+, is analysed for high temperature stability; such an analysis can also provide useful information on the stability of VO2+. Using the information gathered from electrolytes with lower SOCs, the physical state of VO2+ at 100% SOC can be deduced. To demonstrate this strategy, 51V NMR spectra at various temperatures were obtained for a 2.0 M VO2+/0.5 M VO2+ electrolyte (6.5 M total sulfate). As shown in Fig. 3(a), the peak from VO2+ disappeared after 15 °C, leaving behind the peak from VO(OH)3 in the spectra. For the peak from VO(OH)3, the sharper line width and higher chemical shift than those from VO2+ are in good agreement with the results from Fig. 1 and 2. In contrast to the spectra of the 2.0 M VO2+ solution, the VO2+ containing electrolyte more clearly exhibits the temperature dependent evolution of VO(OH)3. This is of practical importance in that the spectroscopic tool provides more reliable assessment of the various approaches to improving the temperature stability. One previous publication suggested that the deprotonation of hydrated VO2+ to form VO(OH)3 occurs above 335 K for a 2.0 M VO2+ solution in 7.0 M total sulfate solution.7 However, the results indicate that, for a highly concentrated vanadium electrolyte, VO(OH)3 can be produced even at room temperature.
 | ||
| Fig. 3 (a) Temperature-dependent 51V NMR spectra for 2.0 M VO2+/0.5 M VO2+/6.5 M total sulfate solution, and (b) plot of line width and chemical shift as a function of temperature. | ||
The changes of the chemical shift and line width with temperature shown in Fig. 3(b) also verify that the abrupt change in chemical shift can be attributed to the disappearance of the VO2+ line due to paramagnetic dipolar broadening. Therefore, the transition in the chemical shift should not be interpreted as a transition from VO2(H2O)3+ to neutral VO(OH)3. In fact, the chemical shift and the line broadening of VO(OH)3 were nearly unchanged with temperature; however, it is notable that the intensity gradually decreased with temperature, probably due to its precipitation in the form of V2O5.
To further demonstrate the applicability of this method, a newly reported electrolyte containing H3PO4 was studied. Recently, it was reported that the addition of 1 wt% H3PO4 to a highly concentrated VO2+ electrolyte can improve the thermal stability of the VO2+ electrolyte.13,20 The authors expected that the H3PO4 may function as a precipitation inhibitor of V2O5. However, a question arises as to whether H3PO4 influences the formation of VO(OH)3, as HCl does.8,9 To answer this question, we applied our NMR technique to a 1 wt% H3PO4 containing vanadium electrolyte. The total vanadium concentration was set at 2.0 M according to a previous publication on the H3PO4 containing electrolyte. Fig. 4 shows the temperature dependence of the H3PO4 containing electrolyte (1 wt% H3PO4 in 1.6 M VO2+/0.4 M VO2+/5.0 M total sulfate). In the NMR spectra measured at different temperatures, the lines from VO(OH)3 were clearly detected. Compared with the spectra for the H3PO4-free electrolytes (shown in Fig. 3(a)), there were no notable differences found. These results indicate that the added H3PO4 may not alter the kinetics of deprotonation of the hydrated VO2+ ion. The chemical shift and the line width of VO(OH)3 for the H3PO4-containing electrolyte (Fig. 4(a)) are nearly the same as those for the H3PO4-free electrolyte (Fig. 4(b)), suggesting that the chemical environments of VO(OH)3 are not different for the two electrolytes.
 | ||
| Fig. 4 (a) Temperature-dependent 51V NMR spectra of 1 wt% H3PO4 added 1.6 M VO2+/0.4 M VO2+/5.0 M total sulfate, and (b) line width and chemical shift as a function of temperature. | ||
As another example of this approach, an ionic organic additive-containing electrolyte was investigated. G. Wang et al. demonstrated that ionic organic additives can improve the high temperature stability of VO2+ electrolyte.11 We conducted a temperature-dependent 51V NMR experiment for 0.5 wt% 3-chloro-2-hydroxypropyl trimethyl ammonium chloride (CHPTAC)-containing electrolyte that showed a pronounced stabilizing effect.11 As shown in Fig. 5, for the CHPTAC-containing electrolyte, the peaks from VO(OH)3 appeared from 288 K as observed for the additive-free electrolyte (Fig. 3). There was no appreciable difference found for the signal from VO(OH)3 between the two electrolytes. However, interestingly, the chemical shift of VO2+ (−589.4 ppm at 288 K) was different from that for the additive-free electrolyte (−579.8 ppm at 288 K). It strongly suggests that, at least, some portion of VO2+ ion interacts with CHPTAC. Since CHPTAC can donate electron to V5+, such up-field shift can be resulted in. It was previously expected that the hydrated VO2+ ions are encapsulated by the additives,11 and the NMR result supports this consideration. The indifference in VO(OH)3 signal between the additive-free electrolyte and the additive-containing electrolyte would be due to the low additive content (0.5 wt%); considering the relative molar quantities of VO2+ and additive, all VO2+ ions may not be stabilized at such low additive content, although a specific interaction exists between VO2+ and additive.
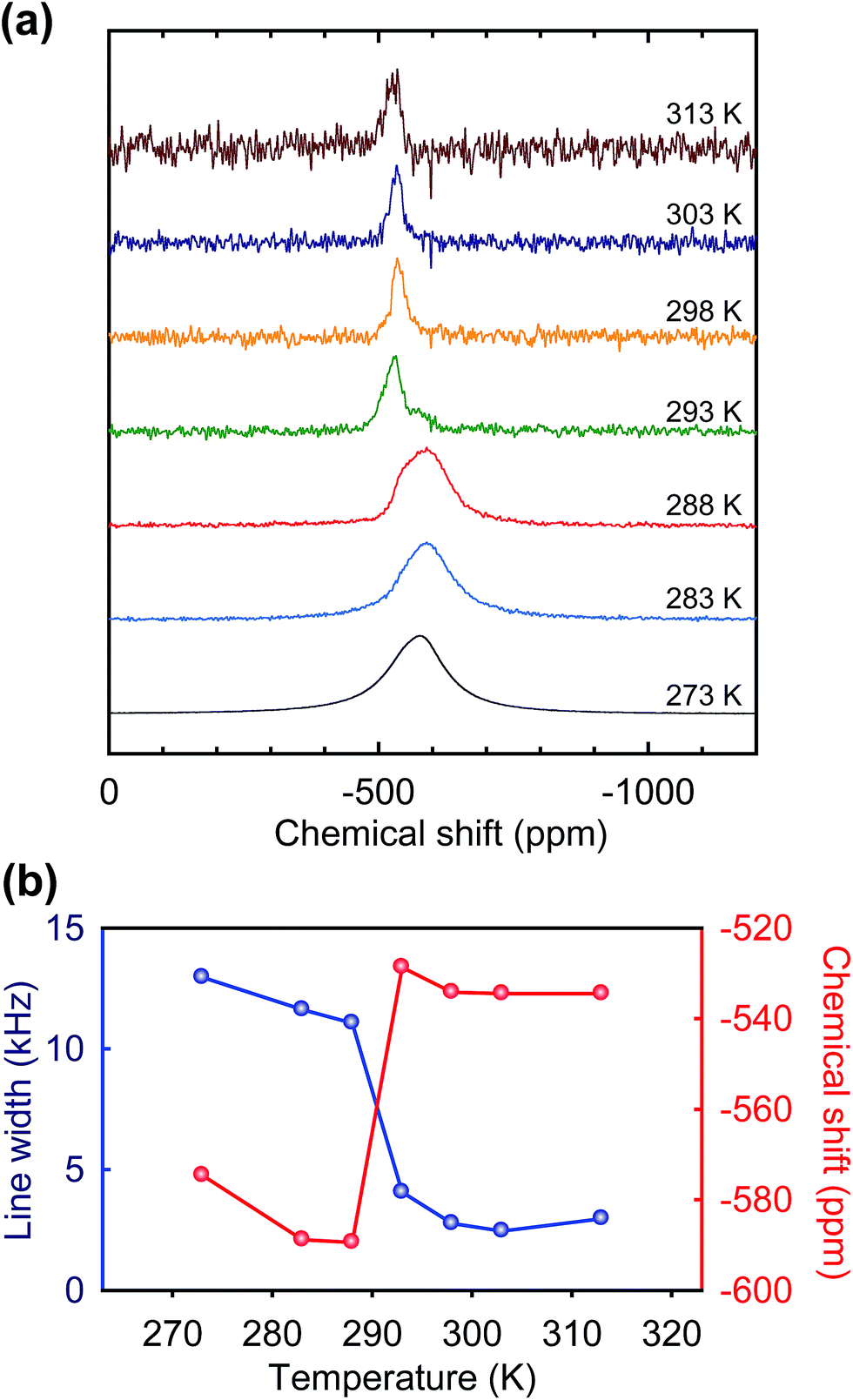 | ||
| Fig. 5 (a) Temperature-dependent 51V NMR spectra of 0.5 wt% CHPTAC added 1.6 M VO2+/0.4 M VO2+/5.0 M total sulfate, and (b) line width and chemical shift as a function of temperature. | ||
4 Conclusions
Paramagnetic dipolar broadening of the VO2+ signal due to a dynamic exchange of VO2+ and VO2+ in a positive vanadium electrolyte was demonstrated. At high temperatures and high acid concentrations, VO2+ can be produced in a VO2+ electrolyte, resulting in significant paramagnetic dipolar broadening. Therefore, in the analysis of the temperature stability of the VO2+ electrolyte with 51V NMR, the influence of paramagnetic dipolar broadening should be carefully considered. By intentionally adding VO2+ in VO2+ electrolyte, a clearer detection of VO(OH)3 was demonstrated. This method indicates that VO(OH)3 can be formed at lower temperatures than previously believed. The induced paramagnetic dipolar broadening can be utilized to assess approaches to improving the temperature stability of the vanadium electrolyte.Acknowledgements
This work was supported by the Korea Institute of Energy Technology Evaluation and Planning (KETEP) and the Ministry of Trade, Industry & Energy (MOTIE) of the Republic of Korea (No. 20142020103710).References
- M. Rychcik and M. Skyllas-Kazacos, J. Power Sources, 1988, 22, 59–67 CrossRef CAS
.
- P. Zhao, H. Zhang, H. Zhou, J. Chen, S. Gao and B. Yi, J. Power Sources, 2006, 162, 1416–1420 CrossRef CAS
- K.-L. Huang, X.-g. Li, S.-q. Liu, N. Tan and L.-q. Chen, Renewable Energy, 2008, 33, 186–192 CrossRef CAS
- M. Skyllas-Kazacos, M. H. Chakrabarti, S. A. Hajimolana, F. S. Mjalli and M. Saleem, J. Electrochem. Soc., 2011, 158, R55–R79 CrossRef CAS
- M. Skyllas-Kazacos, G. Kazacos, G. Poon and H. Verseema, Int. J. Energy Res., 2010, 34, 182–189 CrossRef CAS
- M. Kazacos, M. Cheng and M. Skyllas-Kazacos, J. Appl. Electrochem., 1990, 20, 463–467 CrossRef CAS
- M. Vijayakumar, L. Li, G. Graff, J. Liu, H. Zhang, Z. Yang and J. Z. Hu, J. Power Sources, 2011, 196, 3669–3672 CrossRef CAS
- S. Kim, M. Vijayakumar, W. Wang, J. Zhang, B. Chen, Z. Nie, F. Chen, J. Hu, L. Li and Z. Yang, Phys. Chem. Chem. Phys., 2011, 13, 18186–18193 RSC
- L. Li, S. Kim, W. Wang, M. Vijayakumar, Z. Nie, B. Chen, J. Zhang, G. Xia, J. Hu, G. Graff, J. Liu and Z. Yang, Adv. Energy Mater., 2011, 1, 394–400 CrossRef CAS
- J. Zhang, L. Li, Z. Nie, B. Chen, M. Vijayakumar, S. Kim, W. Wang, B. Schwenzer, J. Liu and Z. Yang, J. Appl. Electrochem., 2011, 41, 1215–1221 CrossRef CAS
- G. Wang, J. Chen, Y. Xu, B. Yan, X. Wang, X. Zhu, Y. Zhang, X. Liu and R. Wang, RSC Adv., 2014, 4, 63025–63035 RSC
- C. Ding, X. Ni, X. Li, X. Xi, X. Han, X. Bao and H. Zhang, Electrochim. Acta, 2015, 164, 307–314 CrossRef CAS
- S. Roe, C. Menictas and M. Skyllas-Kazacos, J. Electrochem. Soc., 2016, 163, A5023–A5028 CrossRef CAS
- B. M. Weckhuysen, Spectroscopy of transition metal ions on surfaces, Leuven University Press, 2000 Search PubMed
- H. M. McConnell and S. B. Berger, J. Chem. Phys., 1957, 27, 230–234 CrossRef CAS
- C. S. Johnson Jr, J. Chem. Phys., 1963, 39, 2111–2114 CrossRef
- R. W. Kreilick and S. Weissman, J. Am. Chem. Soc., 1966, 88, 2645–2652 CrossRef CAS
- J. J. Cruywagen, J. B. B. Heyns and A. N. Westra, Inorg. Chem., 1996, 35, 1556–1559 CrossRef CAS PubMed
- F. Gharib, M. Sayadian, A. Shamel and M. Mobasher-Moghaddam, J. Mol. Liq., 2008, 138, 9–13 CrossRef CAS
- N. Kausar, A. Mousa and M. Skyllas-Kazacos, ChemElectroChem, 2016, 3, 276–282 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2016 |