
Interactions of the N-terminal domain of human islet amyloid polypeptide with lipid membranes: the effect of cholesterol†
Yang Lia,
Liping Guana,
Tong Lua,
Haichao Lib,
Zhengqiang Lib and
Fei Li*a
aState Key Laboratory of Supramolecular Structure and Materials, Jilin University, 2699 Qianjin Avenue, Changchun 130012, P. R. China. E-mail: feili@jlu.edu.cn; Fax: +86-431-85193421; Tel: +86-431-85168548
bKey Laboratory for Molecular Enzymology & Engineering, The Ministry of Education, Jilin University, Changchun 130012, P. R. China
First published on 3rd October 2016
Abstract
The 1–19 region of human islet amyloid polypeptide (hIAPP1–19) is a dominating factor causing the interaction between hIAPP and membrane. It contains a short sequence RLANFLV that fulfils the amino-acid arrangement of the inversed cholesterol recognition amino-acid consensus (CARC) and may mediate a direct contact of hIAPP with cholesterol. In this study, we focused on the interaction of hIAPP1–19 with the lipid membrane composed of 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and cholesterol, and examined the role of the CARC motif in the peptide–membrane interaction. Using differential scanning calorimetry, 31P-NMR spectroscopy, 1H-NMR titration measurement and dye leakage assay, we demonstrated that hIAPP1–19 interacts with DPPC vesicles more strongly in the presence of cholesterol than it does in the absence of cholesterol. The peptide–membrane interaction promotes the domain segregation of the raft-containing membrane. The peptide is more disruptive to the cholesterol-containing membrane than it is to the cholesterol-depleted membrane. The substitution of the residue Phe at position 15 of hIAPP1–19 by Leu leads to a distinct decrease in the peptide–membrane interaction in the presence of cholesterol, but the effect of the residue substitution on the peptide–membrane interaction is very small in the absence of cholesterol. The circular dichroism data indicated that a conversion of the structure from a random coil to an α-helix is induced by cholesterol for both peptides and the structural conversion is more Chol-dependent for the wild-type peptide than the F15L variant. Our findings suggest that cholesterol could facilitate the insertion and aggregation of the N-terminal domain of hIAPP in the membrane, and the phenylalanine in the CARC motif could be involved in the interaction of the N-terminal domain with Chol.
Introduction
Human islet amyloid polypeptide (hIAPP), also called amylin, is a neuropancreatic hormone composed of 37 amino acids.1,2 Its amyloid deposit has been found in the islets of Langerhans of the pancreas of patients affected by Type 2 Diabetes Mellitus (T2DM).2,3 Although the exact mechanism of T2DM is unknown, hIAPP has been linked to the damage and actual death of β-cells in islets.4–6 The interactions between hIAPP and membranes may be a key process in the toxicity of the amyloid peptide to the cells.Cholesterol (Chol) is one of the important compositions in the cellular membranes concerning neurodegenerative diseases.7–10 However, the effects of Chol on the amyloidogenesis are controversial.11 Lines of evidence support the promotion role of Chol in the polymerization of amyloid peptides,12,13 while some studies demonstrate the inhibition effect of Chol on the amyloid fibrillation.14–17 Chol modulates fibrillogenesis either by a direct interaction with protein or by mediating the interactions of protein with other lipid components (e.g. GM1).11
The molecular mechanisms that control the binding of Chol to proteins have been studied extensively. An important category of Chol-associated proteins include one or more specific Chol-binding motif(s). So far two types of Chol-binding motifs are identified. One is referred as CRAC (cholesterol recognition amino-acid consensus) fulfilling an algorithm of (L/V)–X1–5–(Y)–X1–5–(K/R), another is the reversed version of CRAC, named CARC, fulfilling an algorithm of (K/R)–X1–5–(Y/F)–X1–5–(L/V).18–21 The CRAC and CARC motifs have been found in many transmembrane proteins. In most cases, the central aromatic residues of CRAC and CARC appear to play a key role in the protein–Chol interactions, driven by the CH–π interaction.22 Another main category of Chol-associated proteins are found in a broad range of viruses and amyloid proteins.23–28 These proteins include peptide domains that adopt a tilted orientation when they insert in the membrane. Tilted peptides interact with Chol through a geometric complementarity between the apolar domain of cholesterol and the aliphatic residues of the peptide without a requirement of a consensus amino acid motif.18
The N-terminal domain of hIAPP from residues 1 to 19 (hIAPP1–19) is believed to be essential for the binding of hIAPP to membranes.29–34 Unlike the full length peptide, hIAPP1–19 is weakly fibrillogenic whether in solution or in the presence of membranes.31,32 Nevertheless, previous studies have shown that hIAPP1–19 is toxic to both artificial membranes and islet cells.32,35 The N-terminal region of IAPP adopts an α-helical structure in the membranes containing anionic lipids36,37 and a transmembrane topology,38 and is involved in the early stages of oligomerization of the peptide.39
Notably, the N-terminal region of hIAPP contains a triad of amino-acid residues composed of R11, F15 and V17 that constitutes a CARC-like motif. In previous studies, more attentions have been attracted to the aromatic residue F15, and the influences of the residue on the membrane binding and self-assembly of various hIAPP-derived peptides have been explored. The results from the study of hIAPP12–18 showed that the substitutions of F15 by A and L increase the peptide–membrane interaction and affect the aggregation kinetics, but not affect the amyloid formation of the peptide.40 Tu and Raleigh also found the effects of the substitutions at F15 on the fibrillation rate of hIAPP. They related the effects of different F15 substituents on the fibrillation rate to the α-helical propensity of the single site substituted hIAPP.41 Wiltzius and coworkers reported that hIAPP forms a dimer at the early stage of polymerization through the interaction of the helices at residues 8–18 with each other and the aromatic rings of two F15 make an important contact in the dimer formation.42 The N-terminal domain of hIAPP is also found to bind insulin by the interaction between the 7–19 region of hIAPP and the 9–20 region of insulin, and it is suggested that the hIAPP–insulin binding is mediated by the recognition of the aromatic amino acids Phe15 of hIAPP and Tyr16 of insulin.43 Despite having been extensively studied, the interaction of the N-terminal region of IAPP with the membrane in the presence of Chol and the role of the CARC or F15 in the peptide–membrane interaction are unclear. Because of the importance of both the N-terminal region of hIAPP and Chol in the peptide–membrane binding and the amyloidogenesis of hIAPP, elucidating how the N-terminal region of hIAPP interacts with the membrane in the presence of Chol and the effects of the peptide–membrane interaction on the phase behavior of the membrane and the structure of the peptide are significant for disclosure of the mechanism underlying the membrane disruption by hIAPP.
In this study, we used DPPC as a model membrane to examine the interactions of hIAPP1–19 and its F15L variant hIAPP1–19/F15L with the neutral membrane in the absence and presence of Chol. By measuring the binding affinities of the peptides for the membrane, the phase behaviors of the membrane and the conformational conversion of the peptides, we revealed that Chol plays an important role in the binding of hIAPP1–19 to the membrane. By mediating the insertion in the membrane, the formation of the α-helical conformation, and likely the aggregation, Chol enhances the performance of the N-terminal peptide of hIAPP in the disruption of the membrane. Our results showed that the phenylalanine in the CARC motif is involved in the interaction between hIAPP1–19 and Chol.
Results
Effects of Chol on the peptide–membrane interactions
31P-NMR measurements of DPPC SUVs (small unilamellar lipid vesicles) alone and DPPC SUVs containing 15% and 20% Chol were performed at a lipid-to-peptide ratio (L/P) of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in Tris–HCl buffer (pH 7.4). A single peak from the resonance of the phosphorus atom on the head-groups of the lipid vesicles was observed in the spectra. Compared with the 31P-NMR spectrum of DPPC vesicles alone, the signal of DPPC vesicles containing Chol was broadened with increasing Chol concentration (Fig. 1A). When hIAPP1–19 was incorporated into the vesicles, the signal of DPPC vesicles was broadened further either in the absence or in the presence of Chol. However, the signal broadening induced by the peptide was more pronounced in the presence of Chol than that in the absence of Chol. The signal even disappeared at a 20% Chol concentration (Fig. 1B). The incorporation of hIAPP1–19/F15L into DPPC SUVs also resulted in a broadening of the 31P-NMR signals both in the absence and presence of Chol (Fig. 1C). However, the effects of the hIAPP1–19/F15L on the signal of DPPC vesicles were obviously smaller than those of hIAPP1–19 in the presence of Chol, while the effect of the variant on the signal of DPPC vesicles was similar to that of the wild-type peptide in the absence of Chol, suggesting that there may be an interaction between hIAPP1–19 and Chol and F15 may play a role in the peptide–Chol interaction.
:1 in Tris–HCl buffer (pH 7.4). A single peak from the resonance of the phosphorus atom on the head-groups of the lipid vesicles was observed in the spectra. Compared with the 31P-NMR spectrum of DPPC vesicles alone, the signal of DPPC vesicles containing Chol was broadened with increasing Chol concentration (Fig. 1A). When hIAPP1–19 was incorporated into the vesicles, the signal of DPPC vesicles was broadened further either in the absence or in the presence of Chol. However, the signal broadening induced by the peptide was more pronounced in the presence of Chol than that in the absence of Chol. The signal even disappeared at a 20% Chol concentration (Fig. 1B). The incorporation of hIAPP1–19/F15L into DPPC SUVs also resulted in a broadening of the 31P-NMR signals both in the absence and presence of Chol (Fig. 1C). However, the effects of the hIAPP1–19/F15L on the signal of DPPC vesicles were obviously smaller than those of hIAPP1–19 in the presence of Chol, while the effect of the variant on the signal of DPPC vesicles was similar to that of the wild-type peptide in the absence of Chol, suggesting that there may be an interaction between hIAPP1–19 and Chol and F15 may play a role in the peptide–Chol interaction.
 | ||
| Fig. 1 31P-NMR spectra of DPPC SUVs containing various percentages of Chol in the absence (A) and presence of hIAPP1–19 (B) and hIAPP1–19/F15L (C) measured in Tris–HCl buffer at pH 7.4, 50 °C. | ||
The observation of the difference in the interactions of hIAPP1–19 and hIAPP1–19/F15L with the Chol-containing DPPC membrane in the 31P-NMR measurements impelled us to explore the role of Chol in the peptide–membrane interactions further. For this purpose, we performed the 1H-NMR titration experiments and evaluated the peptide–membrane binding affinity. The 1H-NMR signals of the peptides broadened gradually with the increase in the lipid-to-peptide ratio. The intensities of the proton signals at the resonances of Hδ on His18, Hβ on Asn14 and Hη on Arg11 were used to calculate the dissociation constants (ESI†). The dependences of the intensities (I/I0) upon the lipid-to-peptide ratios at various concentrations of Chol were plotted and the dissociation constants (Kd) of the peptide–membrane binding were obtained by the curve fitting (Fig. 2 and Table 1). The data in Fig. 2C and Table 1 showed that the dissociation constants decrease with increasing Chol concentration for the two peptides, and the Kd values of hIAPP1–19 are smaller than those of hIAPP1–19/F15L at the same Chol concentrations. In contrast, the Kd values of the two peptides in the absence of Chol are very close. This indicates that the differences in Kd values of different peptides arise mainly from the interactions between the peptides and Chol, but not the interactions between the peptides and DPPC component. The 1H-NMR results further confirm that Phe15 has a contribution to hIAPP1–19–Chol interaction.
 | ||
| Fig. 2 (A and B) Dependences of the 1H-NMR intensities of the peptides on the lipid-to-peptide ratios at various percentages of Chol: (A) hIAPP1–19 and (B) hIAPP1–19/F15L. (C) The logarithm values of the dissociation constants Kd of the peptides at various Chol concentrations obtained by the fitting of the curves in A and B. The data were obtained in Tris–HCl buffer of the DPPC/Chol SUVs incorporated with the peptides at pH 7.4, 50 °C. | ||
| Peptide | Chol (%) | Kd (mM) |
|---|---|---|
| hIAPP1–19 | 0 | 8.12 ± 0.18 |
| 5 | 2.16 ± 0.25 | |
| 10 | 0.41 ± 0.02 | |
| 15 | 0.21 ± 0.11 | |
| 20 | 0.15 ± 0.07 | |
| 30 | 0.034 ± 0.0011 | |
| hIAPP1–19/F15L | 0 | 8.62 ± 0.37 |
| 5 | 5.23 ± 0.43 | |
| 10 | 2.06 ± 0.95 | |
| 15 | 0.66 ± 0.13 | |
| 20 | 0.37 ± 0.06 | |
| 30 | 0.088 ± 0.0013 |
It should be noted that all the Kd values of F15L variant obtained at various Chol concentrations are smaller than that of the peptide interacting with the Chol-depleted membrane, even though the direct interaction between the peptide and Chol could be reduced or eliminated due to the absence of the aromatic residue. This suggests that in addition to the direct contact, Chol could affect the interaction of hIAPP1–19 with the membrane by other mechanism.
Effects of the peptides on the domain segregation of the Chol-containing DPPC membrane
The thermotropic phase behaviors of DPPC SUVs alone and DPPC SUVs mixed with 15% and 20% Chol in PBS buffer at pH 7.4 were examined by DSC measurements. As described in previous study,44–46 the thermogram of DPPC SUVs alone displayed a pre-transition from a lamellar gel phase (Lβ′) to a rippled gel phase (Pβ′) at 34.5 °C and a main transition from a rippled gel phase to a liquid-crystalline phase (Lα) at 41.6 °C. In the presence of Chol, the pre-transition was eliminated, while the main endothermic transition of DPPC was broadened dramatically. Moreover, an asymmetric profile consisting of two overlapping transitions was observed in the presence of 15% Chol. A deconvolution treatment to the asymmetric profile showed a sharper peak at a lower transition temperature and a broader one at a higher transition temperature. The sharper one was assigned to the melting of Chol-poor domain and another to the melting of Chol-rich domain (Fig. 3A and Table 2). In the presence of 20% Chol, the domain segregation was not clearly observed. Nevertheless, we deconvoluted the DSC endotherm profile using two-component fitting and obtained the thermodynamic parameters as listed in Table 2. | ||
| Fig. 3 DSC thermograms of DPPC vesicles alone and DPPC vesicles containing various quantities of Chol in the absence (A) and presence of hIAPP1–19 (B) and hIAPP1–19/F15L (C). | ||
| System | Chol (%) | Chol-poor domain | Chol-rich domain | ΔHt (kcal mol−1) | ΔTm (°C) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Tm (°C) | ΔT1/2 (°C) | ΔH (kcal mol−1) | Tm (°C) | ΔT1/2 (°C) | ΔH (kcal mol−1) | ||||
| DPPC/Chol | 0 | 41.6 | 0.97 | 6.1 | 6.1 | 0 | |||
| 15 | 40.1 | 2.75 | 2.6 | 41.9 | 4.40 | 1.5 | 4.1 | 1.8 | |
| 20 | 40.4 | 5.77 | 2.5 | 42.9 | 5.70 | 0.9 | 3.4 | 2.6 | |
| DPPC/Chol/hIAPP1–19 | 0 | 40.3 | 1.28 | 5.7 | 5.7 | 0 | |||
| 15 | 39.6 | 1.91 | 2.2 | 42.2 | 4.44 | 2.8 | 5.0 | 2.6 | |
| 20 | 38.3 | 3.15 | 1.3 | 41.4 | 4.41 | 2.2 | 3.5 | 3.1 | |
| DPPC/Chol/hIAPP1–19/F15L | 0 | 41.0 | 1.86 | 5.5 | 5.5 | 0 | |||
| 15 | 38.7 | 1.60 | 2.0 | 40.9 | 5.18 | 2.5 | 4.5 | 2.2 | |
| 20 | 38.6 | 5.45 | 1.7 | 41.5 | 7.12 | 1.3 | 3.0 | 2.9 | |
When hIAPP1–19 was incorporated with DPPC vesicles (Fig. 3B), the pre-transition temperature Tp was decreased by 4.8 °C and the main transition temperature Tm was decreased by 1.3 °C along with the decreases in the cooperativity (ΔT1/2) and the melting enthalpy change (ΔH). This indicates that hIAPP1–19 interacts with DPPC vesicles, by which the lipid chain packing is disturbed. However, when hIAPP1–19 was incorporated with the DPPC vesicles containing 15% and 20% Chol, more distinct separation of the main transition peaks was observed in the DSC profiles (Fig. 3B) compared with the thermogram of DPPC/Chol system without the peptide (Fig. 3A). A deconvolution treatment revealed that the differences between the main transition temperatures (ΔTm) of the Chol-poor and Chol-rich domains are 2.6 °C and 3.1 °C in the presence of 15% and 20% Chol, respectively, which are larger than those of DPPC/Chol systems without the peptide (1.8 and 2.6, respectively, see Table 2). Moreover, the incorporation of hIAPP1–19 with DPPC/Chol membrane led to a decrease in the enthalpy change of the gel to liquid-crystalline phase transition in the Chol-poor domain and an increase in the enthalpy change in the Chol-rich domain compared with the results of the Chol/DPPC systems. This suggests that the incorporation of hIAPP1–19 with the lipid bilayers may induce a redistribution of Chol by mediating more Chol molecules clustering in the Chol-rich domain, and/or induce a more intensive perturbation to the lipid packing in the Chol-poor domain than it does to the Chol-rich domain by selectively interacting with specific regions of the raft-containing membrane.
When hIAPP1–19/F15L was incorporated with DPPC vesicles, the main transition peak was broadened and shifted to a lower Tm by 0.6 °C along with a decrease in the enthalpy change, while the pre-transition temperature was unexpectedly increased by about 1.3 °C (Fig. 3C). The change in Tp may be associated with a change in the ripple periodicity or a change in the membrane curvature induced by the peptide.35 The incorporation of hIAPP1–19/F15L with lipid membrane may decrease the ripple periodicity by inserting in membrane at a proper position, most likely at the position between the head-groups and the aliphatic chains. In the presence of Chol, the effects of hIAPP1–19/F15L on the profiles of the main transitions were small. No obvious differences between the thermograms of the peptide-free and peptide-containing vesicles were observed both at 15% and 20% Chol. However, a deconvolution by a two-peak fitting revealed a peak separation with ΔTm of 2.2 °C (15% Chol) and 2.9 °C (20% Chol) that are smaller than those of DPPC/Chol/hIAPP1–19 systems, but larger than those of DPPC/Chol systems. A decrease in ΔH of the Chol-poor domain and an increase in ΔH of the Chol-rich domain compared with the data of DPPC/Chol vesicles were also observed in the DPPC/Chol/hIAPP1–19/F15L systems. The results suggest that hIAPP1–19/F15L disturbs the distribution of Chol and/or affects the packing of the DPPC/Chol membrane less severely than hIAPP1–19 does.
Effects of Chol on the secondary structures of the peptides
Both hIAPP1–19 and hIAPP1–19/F15L were unstructured in bulk solution, as shown in the CD spectra (Fig. 4A and C). Similar results were also obtained for the peptides incorporated with the Chol-depleted DPPC vesicles. However, when the peptides were incorporated with the Chol-containing DPPC vesicles, a conversion from a random structure to an α-helical structure was observed in the CD spectra of both peptides (Fig. 4B and D). The data obtained by the secondary structure analysis showed that the content of the α-helical structure increases with increasing percentage of Chol, and the increase in the helicity is dramatic from 15% Chol to 20% Chol (Table 3). This suggests that Chol facilitates the formation of α-helix for both peptides. Chol-induced formation of α-helical structure is also observed for Aβ in model membranes.14 Compared with the results of hIAPP1–19, the α-helix contents of hIAPP1–19/F15L were lower at 20% and 30% Chol, implying that the variant inserts in the membrane less deeply and/or aggregates less effectively than the wild-type peptide in the presence of higher percentages of Chol.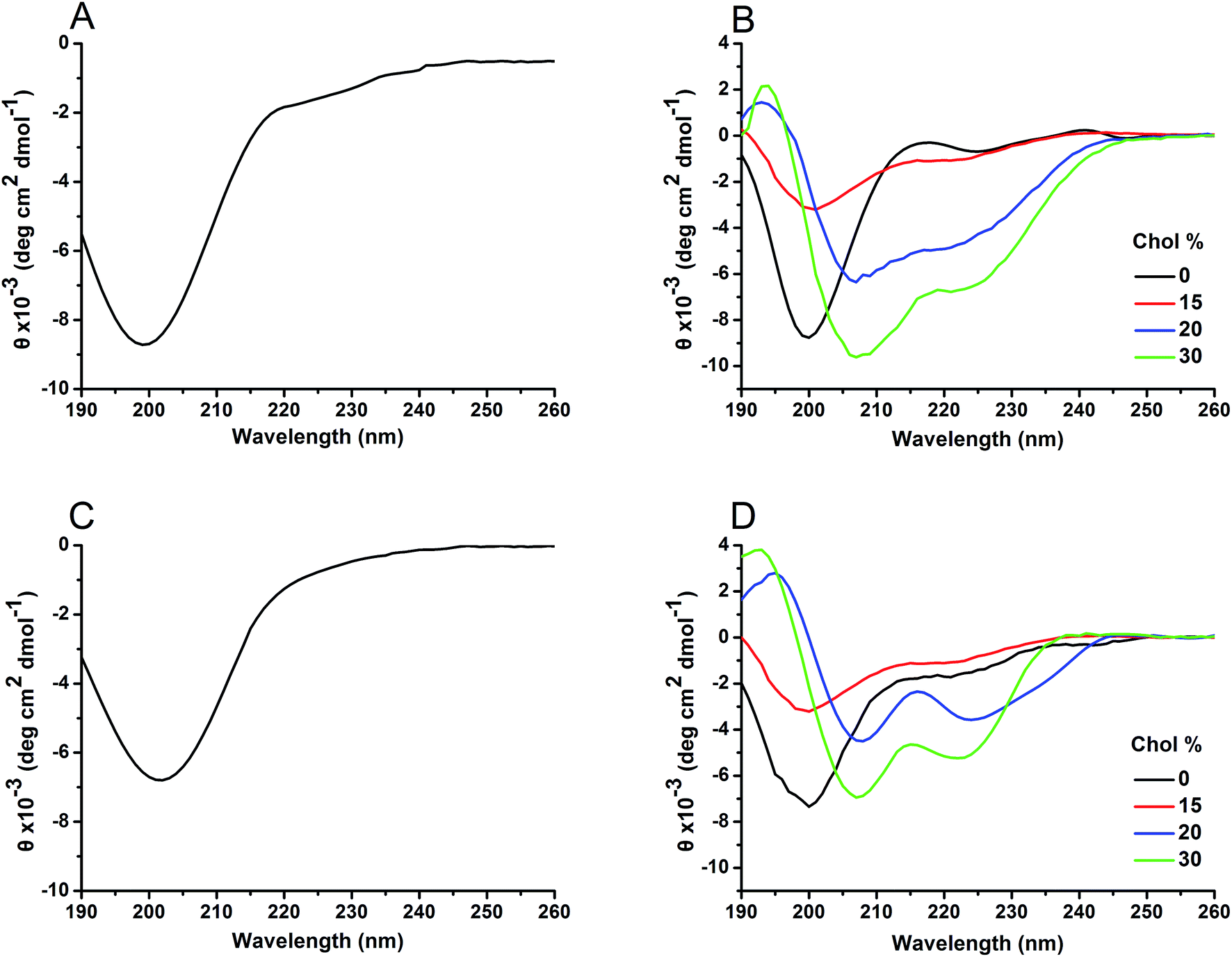 | ||
| Fig. 4 CD spectra of hIAPP1–19 (A) and hIAPP1–19/F15L (C) in PBS and hIAPP1–19 (B) and hIAPP1–19/F15L (D) in DPPC vesicles with various percentages of Chol. | ||
| Peptide | Chol (%) | Secondary structure (%) | |||
|---|---|---|---|---|---|
| Helix | Strand | Turn | Unordered | ||
| hIAPP1–19 | 0 | 18.7 | 18.2 | 14.7 | 48.4 |
| 15 | 24.4 | 17.4 | 16.3 | 41.9 | |
| 20 | 46.4 | 16.2 | 17.1 | 20.3 | |
| 30 | 51.9 | 15.5 | 14.5 | 18.1 | |
| hIAPP1–19/F15L | 0 | 18.9 | 19.5 | 13.8 | 47.8 |
| 15 | 24.0 | 18.9 | 15.8 | 41.3 | |
| 20 | 40.7 | 15.6 | 18.2 | 25.5 | |
| 30 | 45.8 | 16.1 | 15.3 | 22.8 | |
Effect of Chol on the peptide-induced membrane disruption
Previous studies have shown that hIAPP1–19 can disrupt POPG vesicles to a similar extent as full-length IAPP without forming amyloid fibers.32,35 In order to gain insight into the role of Chol in the membrane disruption and the effect of F15L on the activity of the peptide in disrupting membrane, we performed the dye leakage assays of DPPC LUVs (large unilamellar lipid vesicles) in the absence and presence of Chol (Fig. 5). The results demonstrated that both hIAPP1–19 and hIAPP1–19/F15L can disrupt the membranes consisting of DPPC alone and DPPC/Chol mixture. However, the performances of the peptides in the membrane disruption were distinct in the presence and absence of Chol. The peptides induced more amount of dye leakage in the presence of Chol, particularly in the presence of 20% Chol, than they did in the absence of Chol, suggesting a promotion role of Chol in the peptide–lipid interaction and the activities of the peptides in disrupting the lipid membranes. | ||
| Fig. 5 Kinetics of the membrane permeabilization induced by hIAPP1–19 (A) and hIAPP1–19/F15L (B). The peptides of 75 μM were added to calcein containing DPPC/Chol LUVs at the lipid-to-peptide ratio of 20:1. | ||
Moreover, compared with the wild-type peptide, the F15L mutant was less efficient in inducing dye leakage through the membrane. Lines of evidence from previous studies have shown that amyloid peptides disrupt the membrane by their toxic oligomers. Therefore, our results could suggest that Chol promotes the formation of toxic oligomers more efficiently for the wild-type peptide than for the F15L variant.
Discussion
The interactions of the amyloid proteins/peptides with membranes play an important role in the formation of amyloid fibrils and lead to membrane damage.47,48 The insertion of the N-terminal region in membrane, most likely as a monomer, is the initial step of hIAPP–membrane interaction.30 The N-terminal region of hIAPP is also involved in the self-association of hIAPP.39 Because the 1–19 region of hIAPP has a weak fibrillogenic propensity either in solution or at membranes and disrupts lipid membranes similarly to full-length hIAPP, the peptide fragment has been used as a structure mode to investigate the early aggregation and membrane-disrupting mechanism of hIAPP.37 Interestingly, hIAPP1–19 includes a sequence fragment RLANFLV in which the arrangement of amino-acid residues R11, F15 and V17 is consistent with the CARC motif. Therefore, the N-terminal fragment of hIAPP may be also involved in the interaction of the peptide with Chol. In this study, we examined the interactions of hIAPP1–19 with the DPPC membrane in the absence and presence of Chol in order to gain an insight into the mechanisms underlying the effects of Chol on the binding and disruptive activity of hIAPP1–19 to the membranes. The role of CARC in the peptide–membrane interaction was also explored by the substitution of the aromatic residue in CARC by the aliphatic residue leucine.Our results showed that the binding affinity of hIAPP1–19 for the DPPC membrane is very weak (8.12 × 10−3 M), but it increases with the increase in the concentration of Chol (e.g., 3.4 × 10−5 M at 30% Chol). Chol concentration also facilitates the formation of α-helical structure of the peptide, while in the Chol-depleted DPPC membrane, the peptide is unstructured. The structural conversion of the peptide induced by Chol could be elucidated by the topological change of the peptide from lying at membrane surface to inserting into the hydrophobic region of the membrane. In the absence of Chol, the peptide could bind to the surface of the neutral membrane with a lower affinity, which could not induce a defined secondary structure. The presence of Chol leads to the formation of micro-domains in the DPPC membrane, which could facilitate the insertion of hIAPP1–19 in the membrane deeply. Chol is also an important factor affecting the membrane insertion and secondary structure of β-amyloid peptide (Aβ 1–40).14 With the contrary of our result, the study of L Caillon and coworkers demonstrated that Chol does not have any significant effect on the binding of full length hIAPP to DOPC vesicles (the binding affinities are 1.4 × 10−3 M and 2.9 × 10−3 M, respectively, in the absence and presence of 30% Chol).49 The difference in the Chol effect could be partly associated with a stronger aggregation propensity of full length hIAPP than hIAPP1–19 fragment. The pre-oligomerization of full length hIAPP may occur before binding to the membrane, which may impair the interaction of the N-terminal fragment with Chol.
The DSC data demonstrated that the domain segregation associated with Chol heterogeneous distribution in the membrane is enhanced instead of being reduced by the incorporation of hIAPP1–19. This indicates different interactions of hIAPP1–19 with membrane components or domains. The incorporation of hIAPP1–19 with the DPPC membrane containing Chol resulted in a distinct decrease in the melting enthalpy change of the Chol-poor domain and an increase in the melting enthalpy change of the Chol-rich domain, suggesting that the peptide preferentially partitions into the Chol-poor domain of the membrane, alternatively, the peptide renders further clustering of Chol in the membrane. R. Winter and coworkers have evidenced the preferential partitioning of IAPP into the fluid lipid phase (Chol-poor domain) using the membrane systems of both giant unilamellar lipid vesicles (GUVs) of DOPC:DPPC:Chol 1:2:1 and pancreatic INS-1E β-cells.50,51 They suggested that IAPP is mostly absorbed at the rim of the domains. Both the experimental and molecular dynamics simulation results demonstrate that the addition of Chol in PC membranes leads to an increase in the membrane thickness.52,53 The enrichment of Chol in raft domains leads to differences in the thickness of the Chol-rich (lipid-ordered lo) domains compared with the Chol-poor (lipid-disordered ld) domains in the bilayers.54 As a result, there is a significant strain on the boundary between the lo and ld domains, and this boundary region is more vulnerable to membrane disrupting agents.55 The binding in the boundary diminishes energetic costs and relieves the associated tension.54 Therefore, the interaction of hIAPP1–19 with the heterogeneous DPPC membrane may be explained by the preferential insertion of the peptide in the border between the Chol-poor and Chol-rich domains, where Chol promotes the peptide–membrane interaction likely by hydrophobic effects.39,53 The existence of raft induces a curvature strain on membrane, which could also facilitate the binding of peptide to membrane.56
Previous study has showed that Chol stimulates the aggregation of hIAPP into compact 200–500 nm clusters both in the neutral and anionic lipid membranes, while hIAPP forms pore-like globular oligomers with 25–35 nm in size in the membrane without Chol.47 The Chol-induced clustering of peptide could also occur in the DPPC/Chol system of hIAPP1–19. By promoting the formation of toxic oligomers of the peptide, Chol renders the peptide more disruptive to the membrane. Our DSC results showed that the peptide mainly affects the packing of the Chol-poor domains, but less affects the packing of the Chol-rich domains, suggesting that the peptide could accumulate predominantly at the side of the Chol-poor domain of the boundary, but not the side of the raft. The preferential interaction of the peptide with specific regions of the membrane could further restrict the flexibility of the peptide and therefore facilitate the clustering of the peptide (Fig. 6). The results of the dye leakage assays showed that the amount of dye leakage increased with increasing Chol percentage in the DPPC LUVs. This may be attributed to the increases both in the amount of toxic oligomers and in the length of the boundary regions at the higher cholesterol concentration.
 | ||
| Fig. 6 Schematic depiction of the mode of the interaction between hIAPP1–19 and DPPC membrane in the absence and presence of Chol. | ||
The direct interaction between hIAPP1–19 and Chol could occur when the peptide accumulates at the border of the domains. The CARC motif RLANFLV corresponding to the N-terminal 11–17 region of hIAPP may be involved in the peptide–Chol interaction. The in silico studies revealed that Chol binds to CARC motif via the β-face, leaving the α-face exposed.57 The contacts between the α-face derived by the van der Waals interaction may mediate the clustering of Chol to form micro-domain in lipid membrane. The self-recognition properties of Chol in turn favor the oligomerization of the CARC-containing peptide. The residue Phe at position 15 is one of a triad of essential amino acid residues in the CARC motif. Our study indicated that compared with the wild-type hIAPP1–19, the F15L variant has a distinctly lower binding affinity for the Chol-containing DPPC membrane, whereas the binding affinities of the two peptides for the Chol-depleted DPPC membrane are very similar. This suggests that Phe15 may play a role in the interaction of hIAPP1–19 with Chol. The F15L substitution may also decrease the effect of the N-terminal peptide on the lipid-based segregation of Chol and the formation of α-helical structure of the peptide in the Chol-containing membrane. The membrane damage induced by hIAPP1–19 is also reduced by the substitution, even though the extent of the effect seems small. These changes in the performance of the peptide disturbing the membrane property and in the secondary structure of the peptide derived by the F15L substitution may be ascribed partly to the changes in the peptide–Chol interaction.
In the interaction of the peptide with Chol, the aromatic residue in the CARC motif stacks classically onto one of the sterane rings of Chol by CH–π interaction.58 The branched aliphatic residues Leu or Val are required by the need to accommodate the crevices and asperities of the Chol molecules by numerous van der Waals contacts between these residues and Chol.58 The presence of the basic residue Lys or Arg is crucial for accommodating the OH group of Chol at the membrane surface, even if there is no direct interaction between Chol and K/R. Because the CARC motif is typically involved in the Chol-binding sites of the transmembrane domains that are mainly α-helical,18 the presence of the CARC motif in hIAPP may imply the formation of the α-helix conformation at least in the region of 11–17 of hIAPP1–19 and an insertion of hIAPP1–19 in the membrane with the cationic group emerging at the membrane surface and the aromatic and hydrophobic residues contact with the apolar zone of Chol. The NMR data obtained in the presence of DPC-micelles revealed that residue 7–17 in hIAPP1–19 is involved in an α-helix at a neutral pH and buried in the micelles deeply.38 Our CD results showed that the percentage of the α-helical structure in hIAPP1–19 is about 52% in the presence of 30% Chol in the DPPC membrane and the content is smaller at a lower concentration of Chol. This could suggest that the CARC motif is involved in the α-helical structure in the Chol-containing DPPC membrane, considering that the disulfide bond between C2 and C7 prevents the formation of α-helical structure in the N-terminal region of hIAPP1–19. The absence of the aromatic ring in hIAPP1–19/F15L decreases the helical content and membrane insertion of the peptide likely by the decrease or even the elimination of the direct peptide–Chol interaction, leading to the decrease in the effect of the peptide on the membrane.
Experimental section
Materials
The peptides corresponding to the residues from 1 to 19 of hIAPP (hIAPP1–19) and those with a substitution of phenylalanine at position 15 by leucine (hIAPP1–19/F15L) were synthesized by Shanghai Science Peptide Biological Technology Co., Ltd (Shanghai, China). The cysteines at positions 2 and 7 in both peptides were oxidized to form a disulfide bond and the C-termini of the peptides were amidated. The purity of the peptides was assessed to be higher than 95% by high-performance liquid chromatography and mass spectroscopy. Lipid 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) was purchased from Avanti Polar Lipid, Inc. (Alabaster, AL). Other chemical agents were obtained from Sigma-Aldrich (St. Louis, MO). All chemical agents were used directly without further purification.Preparation of small unilamellar lipid vesicles (SUVs) and large unilamellar lipid vesicles (LUVs)
DPPC and Chol powders were separately dissolved in 200 μL chloroform/methanol (2:1 v/v) co-solvent. Appropriate volume of DPPC solution was mixed with certain volume of Chol solution according to desired percentage of Chol in total lipids. The solution mixture was evaporated by a stream of nitrogen, and the resulting lipid film was kept under vacuum overnight to remove residual organic solvents. The dried lipid film was hydrated with buffer solution and vortexed for several seconds. The vesicle suspension was sonicated for 1 h at 10 °C above the phase transition temperature of the lipids, by which the SUVs were obtained. The liposome samples were used immediately after preparation.
To study the dye leakage of membrane, we prepared LUVs containing calcein. The dried lipid film was hydrated with 1 mL Tris–HCl buffer containing 70 mM calcein. To increase solubility of calcein, 5 M NaOH was added until the solution turned transparent. The solution was incubated for 1 h at 50 °C. Then the solution was freeze-thawed 5 times and extruded through polycarbonate filter (0.1 μm pore size) for 10 cycles. To eliminate the nonencapsulated calcein, the LUVs solution was dialyzed in Tris–HCl buffer over night through a membrane with a cut-off of 1000 Da.
Preparation of peptide-SUVs samples
Peptide was dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) at a concentration of 1 mg mL−1. The solution was sonicated in water bath for 15 min to break up any preformed aggregates of peptide. An appropriate volume of peptide stock solution was added in a mixture of DPPC and Chol that were dissolved previously in a chloroform/methanol (2:1 v/v) co-solvent at a certain percentage of Chol. The solution was further treated following the method described above in the preparation of SUVs. By this method, the samples of peptide incorporated with SUVs in buffer solution were obtained.
Differential scanning calorimetry (DSC) measurements
DSC scans were carried out using a Microcal VP-DSC calorimeter (MicroCal, Inc., Northampton, MA). Both the sample and reference were degassed by vacuum for 10 min and heated from 20 to 60 °C with a heating rate of 0.5 °C min−1. To ensure the reproducibility and accuracy of the experiments, three independent samples were measured. The results were analyzed after buffer subtraction and baseline correction using Microcal Origin software. The SUV samples of 1.5 mM DPPC/Chol mixed with 75 μM peptide (L/P = 20:1) dissolved in 25 mM phosphate buffer (pH 7.4) containing 50 mM NaCl were used. The mole percentages of Chol composition in DPPC/Chol mixtures were varied from 0, 15% to 20%.
NMR spectroscopy
1H- and 31P-NMR spectra of the peptide/SUV samples were performed on a Bruker Avance 600 spectrometer (Bruker BioSpin, Fällanden, Switzerland) at 50 °C. The SUV samples of DPPC/Chol and DPPC/Chol/peptide (L/P = 20:1) mixtures dissolved in 10 mM Tris–HCl buffer (pH 7.4) containing 100 mM NaCl and 10% D2O were used in all NMR experiments. 31P spectra were collected with scans of 2592 and a relaxation delay of 1.5 s. The 31P chemical shift of H3PO4 was used as a reference chemical shift. A total of 300 μM peptide and 6 mM lipids (DPPC and Chol) were used in the 31P-NMR experiments.
1H-NMR titration experiments of the peptide incorporated with SUVs were performed at different Chol concentrations: 0, 5, 10, 15, 20 and 30 mol%. By increasing the concentration of lipid component from 0 to 15 mM at a fixed Chol:DPPC ratio and peptide concentration (300 μM), a series of 1H-NMR spectra of peptide in DPPC/Chol SUVs with different lipid-to-peptide ratios were obtained. All spectra were collected with scans of 512 and a relaxation delay of 3 s. DSS (2,2-dimethyl-2-silapentane-5-sulfonate-d6) was used as an internal standard.
The intensities of the peptide signals were measured at different L/P ratios and the dissociation constants (Kd) of the peptide binding to SUVs were estimated by eqn (1) derived from a simple bimolecular binding equilibrium:49,59
 | (1) |
Circular dichroism (CD) spectroscopy
CD measurements were performed on a PMS-450 spectropolarimeter (Biologic, France) at room temperature. Samples used in CD measurements were prepared similar to those used in DSC measurements. A 25 mM phosphate buffer solution (pH 7.4) without salt was used in the preparation of the CD samples. The sample solutions were added into a quartz cuvette with 0.5 mm path length. The spectra were scanned from 190 nm to 260 nm with a step of 1 nm in an interval of 10 s. The background collected from the peptide-free sample was subtracted. Each spectrum represented an average of three independent experiments and a smoothing algorithm was used. The absorbance was expressed as molar ellipticity (θ) in a unit of deg cm2 dmol−1. The concentrations of peptide and lipids were 75 μM and 1.5 mM, respectively.The secondary structure contents were calculated by the CDPro software package using the program CONTIN/LL. A reference set of SMP56 including 56 proteins was used in the analyses of CD data.
Membrane leakage assays
The membrane leakage assays were performed on a fluorescence spectrophotometer RF-5301 PC (Shimadzu, Japan) at room temperature. The fluorescence spectra were excitated at 495 nm with emission wavelength from 530 nm to 650 nm. The spectra were scanned with an excitation slit of 3 nm and an emission slit of 3 nm. Each spectrum was an average of three time scans. The standard deviations were estimated based on three separate measurements. The 100% leakage (I100) was obtained by the calcein-LUVs with 0.2% Triton X-100.The data was analyzed using eqn (2) (ref. 40, 60 and 61):
 | (2) |
Conclusion
Using the raft-containing DPPC/Chol vesicles as a model membrane system, we detected the promotion effects of Chol on the binding affinity of hIAPP1–19 for the membrane and on the disruptive activity of the peptide to the membrane. By the substitution of F to L at position 15, we revealed that the CARC motif composed of a triad of amino acid residues R11, F15 and V17 in hIAPP1–19 plays a role in the peptide–Chol interaction. The peptide variant devoid of the aromatic residue that contacts with the sterane rings of Chol by CH–π interaction binds to the raft-containing membrane more weakly and exerts a smaller effect on the Chol redistribution in the neutral membrane. In addition to the direct interaction of the peptide with Chol, the preferential insertion of the peptide in the boundary region of the Chol-rich and Chol-poor domain in the membrane also plays a role in the Chol-associated peptide–membrane interaction. Chol induces the conversion of the secondary structure of the peptide from a random coil to an α-helix. By exerting specific conformational effects on the peptide in the membrane, Chol could restrict the flexibility of the peptide and result in the formation of the toxic oligomers.Acknowledgements
We thank Dr Chunyu Wang for the help in NMR experiments.Notes and references
- G. J. S. Cooper, A. C. Willis, A. Clark, R. C. Turner, R. B. Sim and K. B. M. Reid, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 8628–8632 CrossRef CAS.
- P. Westermark, C. Wernstedt, E. Wilander, D. W. Hayden, T. D. O'Brien and K. H. Johnson, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 3881–3885 CrossRef CAS.
- E. Gazit, Angew. Chem., Int. Ed., 2002, 41, 257–259 CrossRef CAS.
- C. J. Rhodes, Science, 2005, 307, 380–384 CrossRef CAS PubMed.
- R. L. Hull, G. T. Westermark, P. Westermark and S. E. Kahn, J. Clin. Endocrinol. Metab., 2004, 89, 3629–3643 CrossRef CAS PubMed.
- S. E. Kahn, S. Andrikopoulos and C. B. Verchere, Diabetes, 1999, 48, 241–253 CrossRef CAS PubMed.
- I. J. Martins, T. Berger, M. J. Sharman, G. Verdile, S. J. Fuller and R. N. Martins, J. Neurochem., 2009, 111, 1275–1308 CrossRef CAS PubMed.
- R. A. G. Hu, P. Jousilahti, M. Kivipelto and J. Tuomilehto, Neurology, 2008, 70, 1972–1979 CrossRef PubMed.
- J.-P. Liu, Y. Tang, S. Zhou, B. H. Toh, C. McLean and H. Li, Mol. Cell. Neurosci., 2010, 43, 33–42 CrossRef CAS PubMed.
- L. A. Shobab, G.-Y. R. Hsiung and H. H. Feldman, Lancet Neurol., 2005, 4, 841–852 CrossRef CAS PubMed.
- K. Yanagisawa, Subcell. Biochem., 2005, 38, 179–202 CAS.
- J. R. Harris, Micron, 2002, 33, 609–626 CrossRef CAS PubMed.
- D. A. Bosco, D. M. Fowler, Q. Zhang, J. Nieva, E. T. Powers, P. Wentworth, R. A. Lerner and J. W. Kelly, Nat. Chem. Biol., 2006, 2, 249–253 CrossRef CAS PubMed.
- S.-R. Ji, Y. Wu and S.-F. Sui, J. Biol. Chem., 2002, 277, 6273–6279 CrossRef CAS PubMed.
- S. Micelli, D. Meleleo, V. Picciarelli and E. Gallucci, Biophys. J., 2004, 86, 2231–2237 CrossRef CAS PubMed.
- W.-J. Cho, S. Trikha and A. M. Jeremic, J. Mol. Biol., 2009, 393, 765–775 CrossRef CAS PubMed.
- W.-J. Cho, B. P. Jena and A. M. Jeremic, Methods Cell Biol., 2008, 90, 267–286 CrossRef CAS PubMed.
- J. Fantini, C. D. Scala, C. J. Baier and F. J. Barrantes, Chem. Phys. Lipids, 2016, 199, 52–60 CrossRef CAS PubMed.
- H. Li and V. Papadopoulos, Endocrinology, 1998, 139, 4991–4997 CAS.
- H. Li, Z. Yao, B. Degenhardt, G. Teper and V. Papadopoulos, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 1267–1272 CrossRef CAS.
- C. J. Baier, J. Fantini and F. J. Barrantes, Sci. Rep., 2011, 69, 1–7 Search PubMed.
- J. Fantini and F. J. Barrantes, Front. Physiol., 2013, 4, 1–9 Search PubMed.
- R. Brasseur, T. Pillot, L. Lins, J. Vandekerckhove and M. Rosseneu, Trends Biochem. Sci., 1997, 22, 167–171 CrossRef CAS PubMed.
- R. Brasseur, Mol. Membr. Biol., 2009, 17, 31–40 CrossRef.
- C. D. Scala, N. Yahi, C. Lelièvre, N. Garmy, H. Chahinian and J. Fantini, ACS Chem. Neurosci., 2013, 4, 509–517 CrossRef PubMed.
- C. D. Scala, H. Chahinian, N. Yahi, N. Garmy and J. Fantini, Biochemistry, 2014, 53, 4489–4502 CrossRef PubMed.
- C. D. Scala, J.-D. Troadec, C. Lelièvre, N. Garmy, J. Fantini and H. Chahinian, J. Neurochem., 2014, 128, 186–195 CrossRef PubMed.
- J. Fantini, D. Carlus and N. Yahi, Biochim. Biophys. Acta, 2011, 1808, 2343–2351 CrossRef CAS PubMed.
- W. Xu, G. Wei, H. Su, L. Nordenskiöld and Y. Mu, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 84, 051922 CrossRef PubMed.
- M. F. M. Engel, H. Yigittop, R. C. Elgersma, D. T. S. Rijkers, R. M. J. Liskamp, B. de Kruijff, J. W. M. Höppener and J. A. Killian, J. Mol. Biol., 2006, 356, 783–789 CrossRef CAS PubMed.
- L. Khemtémourian, M. F. M. Engel, R. M. J. Liskamp, J. W. M. Höppener and J. A. Killian, Biochim. Biophys. Acta, 2010, 1798, 1805–1811 CrossRef PubMed.
- J. R. Brender, E. L. Lee, M. A. Cavitt, A. Gafni, D. G. Steel and A. Ramamoorthy, J. Am. Chem. Soc., 2008, 130, 6424–6429 CrossRef CAS PubMed.
- A. M. Ruschak and A. D. Miranker, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12341–12346 CrossRef CAS PubMed.
- C. Goldsbury, K. Goldie, J. Pellaud, J. Seelig, P. Frey, S. A. Müller, J. Kistler, G. J. S. Cooper and U. Aebi, J. Struct. Biol., 2000, 130, 352–362 CrossRef CAS PubMed.
- J. R. Brender, K. Hartman, K. R. Reid, R. T. Kennedy and A. Ramamoorthy, Biochemistry, 2008, 47, 12680–12688 CrossRef CAS PubMed.
- S. A. Jayasinghe and R. Langen, Biochemistry, 2005, 44, 12113–12119 CrossRef CAS PubMed.
- J. D. Knight, J. A. Hebda and A. D. Miranker, Biochemistry, 2006, 45, 9496–9508 CrossRef CAS PubMed.
- R. P. R. Nanga, J. R. Brender, J. Xu, G. Veglia and A. Ramamoorthy, Biochemistry, 2008, 47, 12689–12697 CrossRef CAS PubMed.
- Y. Mazor, S. Gilead, I. Benhar and E. Gazit, J. Mol. Biol., 2002, 322, 1013–1024 CrossRef CAS PubMed.
- D. Milardi, M. F. M. Sciacca, M. Pappalardo, D. M. Grasso and C. L. Rosa, Eur. Biophys. J., 2011, 40, 1–12 CrossRef CAS PubMed.
- L.-H. Tu and D. P. Raleigh, Biochemistry, 2013, 52, 333–342 CrossRef CAS PubMed.
- J. J. W. Wiltzius, S. A. Sievers, M. R. Sawaya and D. Eisenberg, Protein Sci., 2009, 18, 1521–1530 CrossRef CAS PubMed.
- S. Gilead, H. Wolfenson and E. Gazit, Angew. Chem., Int. Ed., 2006, 45, 6476–6480 CrossRef CAS PubMed.
- G. Yang, H. Xu, Z. Li and F. Li, Biochim. Biophys. Acta, 2014, 1838, 2588–2599 CrossRef CAS PubMed.
- D. A. Mannock, R. N. A. H. Lewis and R. N. McElhaney, Biophys. J., 2006, 91, 3327–3340 CrossRef CAS PubMed.
- M. G. K. Benesch, D. A. Mannock, R. N. A. H. Lewis and R. N. McElhaney, Biochemistry, 2011, 50, 9982–9997 CrossRef CAS PubMed.
- L. Khemtémourian, J. A. Killian, J. W. M. Höppener and M. F. M. Engel, Exp. Diabetes Res., 2008, 2008, 421287–421295 CrossRef PubMed.
- S. A. Jayasinghe and R. Langen, Biochim. Biophys. Acta, 2007, 1768, 2002–2009 CrossRef CAS PubMed.
- L. Caillon, L. Duma, O. Lequin and L. Khemtemourian, Mol. Membr. Biol., 2014, 31, 239–249 CrossRef CAS PubMed.
- D. Radovan, N. Opitz and R. Winter, FEBS Lett., 2009, 583, 1439–1445 CrossRef CAS PubMed.
- K. Weise, D. Radovan, A. Gohlke, N. Opitz and R. Winter, ChemBioChem, 2010, 11, 1280–1290 CrossRef CAS PubMed.
- A. Léonard, C. Escrive, M. Laguerre, E. P. Peyroula, W. Néri, T. Pott, J. Katsaras and E. J. Dufourc, Langmuir, 2001, 17, 2019–2030 CrossRef.
- L. S. Vermeer, B. L. de Groot, V. Réat, A. Milon and J. Czaplicki, Eur. Biophys. J., 2007, 36, 919–931 CrossRef CAS PubMed.
- S. L. Veatch and S. L. Keller, Phys. Rev. Lett., 2002, 89, 268101–268104 CrossRef PubMed.
- M. F. M. Sciacca, F. Lolicato, G. D. Mauro, D. Milardi, L. D'Urso, C. Satriano, A. Ramamoorthy and C. L. Rosa, Biophys. J., 2016, 111, 140–151 CrossRef CAS PubMed.
- P. E. S. Smith, J. R. Brender and A. Ramamoorthy, J. Am. Chem. Soc., 2009, 131, 4470–4478 CrossRef CAS PubMed.
- J. Fantini, C. D. Scala, L. S. Evans, P. T. F. Williamson and F. J. Barrantes, Sci. Rep., 2016, 6, 21907–21920 CrossRef CAS PubMed.
- J. Fantini and F. J. Barrantes, Biochim. Biophys. Acta, 2009, 1788, 2345–2361 CrossRef CAS PubMed.
- C. M. Pfefferkorn and J. C. Lee, J. Phys. Chem. B, 2010, 114, 4615–4622 CrossRef CAS PubMed.
- C. A. D. Carufel, N. Quittot, P. T. Nguyen and S. Bourgault, Angew. Chem., Int. Ed., 2015, 54, 14383–14387 CrossRef PubMed.
- M. F. M. Engel, L. Khemtémourian, C. C. Kleijer, H. J. D. Meeldijk, J. Jacobs, A. J. Verkleij, B. de Kruijff, J. A. Killian and J. W. M. Höppener, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6033–6038 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra19714k |
| This journal is © The Royal Society of Chemistry 2016 |