DOI:
10.1039/C6RA20484H
(Paper)
RSC Adv., 2016,
6, 99859-99866
A phenanthroline-derived ligand and its complexation with Pd(II): from ligand design, synthesis and Pd(II) complexes structures to its application†
Received
14th August 2016
, Accepted 29th September 2016
First published on 30th September 2016
Abstract
A novel phenanthroline-derived bis-opened-triazine ligand for selective complexation with Pd(II) over 19 typical metals from HNO3 media was designed based on cavity modulation strategy. Structures of the three species of the Pd(II) complexes of with the ligand were elucidated by 1H NMR titration isothermal titration calorimetry (ITC) and density functional theory (DFT) studies. ITC test and binding energy calculations demonstrated that the asymmetrical 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Pd(II)–ligand complex with a monodentate nitrate anion was the most stable among these three species of the Pd(II) complexes. The excellent kinetics of Pd(II) extraction with the ligand is due to its optimal conformation completely ready for metal ligation. The enhanced special extraction selectivity and fast extraction rate towards Pd(II) achieved by the phenanthroline-derived ligand demonstrated that the effective separation of some radionuclides with smaller radius in HLW through cavity modulation strategy is a feasible approach.
:1 Pd(II)–ligand complex with a monodentate nitrate anion was the most stable among these three species of the Pd(II) complexes. The excellent kinetics of Pd(II) extraction with the ligand is due to its optimal conformation completely ready for metal ligation. The enhanced special extraction selectivity and fast extraction rate towards Pd(II) achieved by the phenanthroline-derived ligand demonstrated that the effective separation of some radionuclides with smaller radius in HLW through cavity modulation strategy is a feasible approach.
Introduction
Highly active liquid waste (HLW) generated from Purex (plutonium and uranium reduction extraction) process which is used to extract most of the major uranium and plutonium from nuclear waste, contains the long-lived minor actinides (such as Am and Cm), Pd, Zr, I, Tc, and other short-lived fission or corrosion products.1 The promising technique ADS (Accelerator Driven Sub-critical System) involving the partitioning and transmutation of those long-lived radionuclides from HLW has been thoroughly investigated to minimize the long-termed radiotoxicity of the radioactive wastes to the environment for the last two decades.2 The development of some satisfying organic ligands capable of selectively complexing and extracting the long-lived radionuclides such as Am, Cm and 107Pd, etc. out of the other fission or corrosion products,3 especially those with high neutron capture cross sections such as lanthanides, plays a key role in ADS.4
For Am and Cm, two important ligands were CyMe4-BTPhen 1 (ref. 3b) and CyMe4-BTBP 2 (ref. 5) as shown in Fig. 1, exhibiting high extraction selectivity for minor actinides over lanthanides. The very recently exploited ligand 1 had a more efficient separation capability for Am(III) than the early developed ligand 2, as was the currently used reference extractant of SANEX (Selective Actinide Extraction) process in Europe.6
 |
| | Fig. 1 Molecular structures as well as the coordination cavities of CyMe4-BTPhen, CyMe4-BTBP, and CA-BOPhen. | |
For 107Pd(II), however, little research has been focused on its separation from HLW. The total content of the long-lived 107Pd(II) (t1/2 = 6.5 × 106 years) along with the other short-lived palladium isotopes in HLW is about 1.0–2.0 kg t−1, even higher than that of the aforementioned actinides.7 Being a typical Lewis soft acid, Pd(II) can form stable complexes with some soft S/N-donor ligands. Therefore various S or N-donor ligands have been investigated for the separation and recovery of Pd(II) from HLW.8 However, S containing ligands will inevitably give rise to the secondary waste, which violated the “CHON principle” and limited their practical applications in process of the nuclear waste management.9 Although some N-donor ligands like (methylimino) bis(N,N-dioctylacetamide) MIDOA10 or dicarboxypyridine diamide (DCPDA)11 have been developed for the extraction of Pd(II) from highly acidic HNO3 solution, unfortunately some other metal ions were also co-extracted by these ligands. Until now no N-donor ligands have been reported that have the ability to extract toward Pd(II) over all other fission or corrosion products with high selectivity and fast extraction kinetics.8b Thus, highly efficient soft N-donor extractants without presence of sulfur are valuable to be developed for selective separation and recovery of Pd(II) from HLW.
Recently, it was reported that phenanthroline and bipyridine-derived quadridentate bis-triazine ligands were easily coordinated with lanthanides to predominantly form the stable 1:2 bis-complexes. According to single crystal X-ray crystallography and theoretical calculation, the radius of coordination cavity formed by two ligand molecules together with an extra nitrate anion, are estimated to be among 2.50–2.65 Å.3d,12 For these bis-triazine ligands, geometrically, the nicety match between the size of coordination cavities and the radius of minor actinides (MAs) may be responsible for their excellent extraction performances toward MAs (Am(III) and Cm(III)). In addition, the rigidity of planar skeleton structure with proper geometry and distances of the four nitrogen atoms endeavor to a concrete cavity suitable for selective recognition of the Am(III).
In comparison, the Pd(II) ion with a smaller ionic radius than that of Am(III), usually forms the four-coordinate square-planar geometric complexes,13 although six coordinate species are occasionally encountered14. Nicely, these phenanthroline and bipyridine-derived bis-triazine ligands are four coordinate nitrogen heterocyclic structure, which make it easier to complex with Pd(II). Taking into these considerations, herein, we presented a novel phenanthroline-based N-donor ligand CA-BOPhen 3 (Fig. 1) with triazine ring opened, instead of triazine ring closed planar counterpart in 1 and 2. Molecules of 3 could build a relatively narrow cavity taking advantages of the flexible acyclic and the two amino groups, rightly match the smaller Pd(II). However, to the best of our knowledge, there is no research that has been reported on the complex composition, structure as well as the separation behavior of Pd(II) with these phenanthroline-derived ligands. This is a case for enhancing the extraction selectivity through changing the side chain moieties of phenanthroline ligands to build a smaller cavity suitable for radionuclides with smaller radius in HLW.
Experimental section
Materials and methods
Commercially available organic and inorganic reagents were purchased in China and used as received without further purification except for the anhydrous ether which was dried by P2O5 and zeolite. NMR spectra were recoded with a Bruker Avance 500 spectrophotometer or a Bruker Avance DMX 400 spectrophotometer. ESI-MS were taken on a Bruker Esquire 3000 Plus spectrometer. FT-IR spectra were carried out on a Bruker Vector 22 FT-IR spectrometer. DSC-TGA curves were collected on TA SDTQ600 thermal analyzer. Elemental analyses were performed with a Thermo Finnigan Italia S.P.A EA 1112 CHNS elemental analyzer. The concentration values of all the tested metals in aqueous phases were determined by Varian 730-ES ICP-OES or Varian AA 240 Atomic Absorption Spectrometer.
Synthesis and characterization of ligand (CA-BOPhen 3)
Synthesis of 2,9-dicarbaldehyde-1,10-phenanthroline (a)3b. SeO2 (14.0 g, 126.1 mmol) was dissolved in a mixture of dioxane (250.0 mL) and deionized water (12.0 mL), then stirred and heated to reflux. To this mixture was added dropwise by a solution of 2,9-dimethyl-1,10-phenanthroline (10.0 g) dissolved in dioxane (250.0 mL) for over 20 min. Then the mixture was heated under reflux for another 2 hours. The resulting mixture was filtered quickly while hot to remove the solid selenium. The filtrate obtained was cooled enough to get the solid and then washed with ether (3 × 30.0 mL) and dried in a vacuum drying oven to get the product a as a brown solid (9.9 g, 72%).
Synthesis of 2,9-dicarbonitrile-1,10-phenanthroline (b)3b. A mixture of a (9.6 g, 40.8 mmol), hydroxylamine hydrochloride (6.1 g, 2.2 eq.), triethylamine (34.3 mL 6.0 eq.) and acetonitrile (450.0 mL) was placed in a 500 mL of one-necked flask and heated under reflux for 4 h, then cooled to room temperature. P-Toluenesulfonyl chloride (24.7 g, 3.3 eq.) and pyridine (20.6 mL, 6.3 eq.) were added to the above mixture and then heated under reflux for another 32 h. The final reaction mixture was filtered while hot and the solid residue obtained was washed with hot acetonitrile. After removing the solvent by vacuum evaporation, the crude product was obtained and washed with methanol twice (200.0 mL × 2) and diethyl ether (200.0 mL) to obtain the compound b as a light brown solid. Combined the above solid residue and dried in air, a total of 4.0 g of b was collected (yield, 43%).
Synthesis of 2,9-dicarbohydrazonamide-1,10-phenanthroline (c)3b. A mixture of b (3.88 g, 16.9 mmol), hydrazine hydrate (91.0 mL, 98%) and ethanol (250.0 mL) was placed in a 500 mL of one-necked flask and stirred under reflux over 10 days. The resulting reaction mixture was evaporated under reduced pressure to afford a semi-solid which was then triturated by diethyl ether (200.0 mL) to afford the compound c as a brown solid (3.5 g, 71%).
Synthesis of bis-(4,7,7-trimethyl-3-oxobicyclo[2.2.1]-heptan-2-ylidene)-1,10-phenanthroline-2,9-bis(carbohydrazonamid) (CA-BOPhen 3). The compound c (3.5 g, 11.8 mmol) was reacted with camphor-quinone d (4.5 g, 2.3 eq.) in a mixture of Et3N (24.0 mL) and THF (250.0 mL) at 68 °C under N2 atmosphere over 7 days. After cooling to room temperature, the resulting mixture was filtered and the solid residue was washed with dichloromethane (40.0 mL). The filtrate was evaporated to get the final residue which was purified by chromatography technology (ethyl acetate/petroleum ether, 1/2) to afford the ligand 3 as a golden yellow solid (1.3 g, 20%). Mp 204–206 °C (recrystallized from EtOH). Found: C, 66.80; H, 6.44; N, 18.12%; C34H38O2N8 requires C, 67.10; H, 6.57; N, 18.42%. FT-IR vmax (KBr, v/cm−1): 3460–3350 (–NH2), 2950, 2854, 1745 (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1630, 1590, 1510, 1500, 1390, 1315, 1250, 1170, 1060, 1000, 939, 889, 864, 746, 696, 634 cm−1. 1H NMR (400 MHz, CDCl3, 298 K) δ (ppm): 8.83 (d, J = 8.3 Hz, 2H), 8.42 (d, J = 8.4 Hz, 2H), 8.03 (s, 2H), 3.54 (d, J = 3.6 Hz, 2H), 2.04 (d, J = 5.1 Hz, 4H, disappeared on D2O shake, H2O), 1.74 (s, 4H), 1.49 (d, J = 5.2 Hz, 4H), 0.99 (s, 12H), 0.83 (s, 6H). 13C NMR (101 MHz, CDCl3, 298 K) δ (ppm):207.62, 165.30, 158.62, 150.47, 144.31, 137.08, 130.81, 127.91, 122.30, 58.76, 48.68, 44.72, 30.88, 24.20, 20.97, 17.87 and 9.12. ESI-MS: m/z = 591.6 [M + H]+, 613.5 [M + Na]+.
O), 1630, 1590, 1510, 1500, 1390, 1315, 1250, 1170, 1060, 1000, 939, 889, 864, 746, 696, 634 cm−1. 1H NMR (400 MHz, CDCl3, 298 K) δ (ppm): 8.83 (d, J = 8.3 Hz, 2H), 8.42 (d, J = 8.4 Hz, 2H), 8.03 (s, 2H), 3.54 (d, J = 3.6 Hz, 2H), 2.04 (d, J = 5.1 Hz, 4H, disappeared on D2O shake, H2O), 1.74 (s, 4H), 1.49 (d, J = 5.2 Hz, 4H), 0.99 (s, 12H), 0.83 (s, 6H). 13C NMR (101 MHz, CDCl3, 298 K) δ (ppm):207.62, 165.30, 158.62, 150.47, 144.31, 137.08, 130.81, 127.91, 122.30, 58.76, 48.68, 44.72, 30.88, 24.20, 20.97, 17.87 and 9.12. ESI-MS: m/z = 591.6 [M + H]+, 613.5 [M + Na]+.
NMR titration experiments
1H NMR titrations of palladium nitrate into a 3 solution were performed to get an insight into the complexation behavior of the Pd(II) with phenanthroline-derived ligands in solution. Due to the poor solubility of ligand 3 in CD3CN, both the solid 3 and Pd(NO3)2·H2O were dissolved in a 3:7 (v/v) mixture of CDCl3 and CD3CN solution, separately. For the studies of Pd(NO3)2·H2O titrated with 3, a 0.5 mL initial solution of 3 (5.3 mM) was added with a solution of Pd(II) (5.3 mM). Conversely, for the studies of ligand 3 titrated into Pd(II), a 0.5 mL Pd(II) (4.5 mM) initial solution was added with the solution of 3 (4.5 mM). After each titration, the solution of the mixture was thoroughly mixed by a minishaker instrument (IKA MS2 Model) for 5 min before the spectra of the ligand were collected. The preliminary contact time experiments demonstrated that this mixing time was sufficient to allow the complexation reaction of Pd(II) with the ligand to reach the equilibrium.
Quantum mechanical (QM) calculations
All the quantum mechanical (QM) calculations were performed by density functional theory (DFT) method in Gaussian 09 package. Geometries of ligand 3, [Pd(3)NO3]+, [Pd(3)2]2+ and [Pd(3)]2+ were fully optimized using hybrid function B3LYP. The split-valence d-polarized 6-311G(d) basis set was used for carbon, hydrogen, oxygen and nitrogen, and effective core potential and triple-split-valence CEP-121G basis set was used for palladium. 13C and 1H NMR spectra for the optimized geometries were calculated at the B3LYP/6311++dp-CEP-121g by using Gauge-Independent Atomic Orbital (GIAO) method. The binding energies for [Pd(3)NO3]+, [Pd(3)2]2+ and [Pd(3)]2+ were calculated at B3LYP/6-311G(d)-CEP-121g, and the counterpoise corrections were performed to remove the basis set superposition error.15,16 Complete Citation of Gaussian Program:17
Isothermal titration calorimetry (ITC) test
ITC experiments were carried out using VP-ITC Micro-calorimeter (Microcal, USA) to determine the association constants (Ka) and some important thermodynamic parameters (enthalpy ΔH°, free energy ΔG°, and entropy ΔS°) of the complexation between Pd(II) and ligand 3 in CH3CN solution (T = 298 K, I = 0.05 M (Et4)NNO3).
Solvent extraction experiments
Equal vols (5.0 cm3) of HNO3 solution containing 5.0 × 10−4 M of 19 typical metals such as alkali metals: Li(I), Na(I), K(I) Rb(I) Cs(I); alkaline earth metals: Mg(II), Ca(II), Sr(II), Ba(II); transition metals: Fe(III), Co(II), Ni(II), Zr(IV), Ru(III) and rare earth elements: La(III), Nd(III), Yb(III), and Y(III) and of n-octanol containing 2.0 mM 3, pre-equilibrated with HNO3 of the same concentration without containing any tested metals, were shaken mechanically in ground glass-stoppered tubes at 298 K with a TAITEC MM-10 Model thermo stated water bath shaker. Preliminary studies showed that the equilibrium was established within 5 min. To ensure that the extraction equilibrium was fully reached, the contact time of two phases was extended to 120 min. The phases were centrifuged and separated. The concentrations of metals in aqueous phases before and after the extraction were determined by the ICP-OES or the AA-240 atomic absorption spectrometer (only for alkali metals). The distribution ratio was calculated as the ratio between the concentrations of metals in the organic phase and the aqueous one. The distribution ratios (DM) of metals were calculated as follows:
where C0 and Ce stand for the initial and equilibrium concentrations of the tested elements in aqueous phase, respectively. The separation factor SFPd/M between Pd(II) and other metals was determined by the ratio between the DM of the corresponding elements:
Results and discussions
Synthesis and ligand structure determination
The novel ligand 3 was synthesized as Scheme 1 shown. Seven days reflux of compound c and another precursor camphor-quinone d in THF under the existence of Et3N led to the target compound 3 with opened triazine moiety instead of planar hexagonal triazine in 1, which may be accounted to the steric hindrance of the near bridge methyl groups. The ligand 3 was characterized by element analysis, FT-IR, NMR, and ESI-MS (Fig. S1–S4 in ESI†). 1H and 13C NMR spectra both suggested the two-fold symmetry of ligand 3 and further, the DFT calculation confirmed the C2 symmetry of 3. The computational patterns and chemical shifts of 1H and 13C NMR spectra of 3 were in good agreements with those of experimental results (Fig. S7b and c, ESI†), respectively.
 |
| | Scheme 1 Synthetic route to CA-BOPhen. | |
1H NMR titration and DFT studies of Pd(II) complexes
The complexation between 3 and Pd(II) was investigated using routine 1H NMR titration by step additions of Pd(NO3)2 into the ligand in a mixture solution of CDCl3 and CD3CN. As shown in Fig. 2, sequential 1H NMR spectra were collected with the increase of M/L (ratio of metal to ligand) from 0.2 to 2.4. Three broad bands around 8.3, 8.8, and 9.0 ppm emerged at the point of M/L = 0.2, growing to maximum around M/L = 1.0–1.2, then decreased gradually to minimum while M/L ≥ 2.0, suggesting that the 1:2 complex between ligand and Pd(II) might be produced.18 Contrasted to the clearly resolved peaks produced by diamagnetic lanthanide complexes previously reported, these broad unresolved peaks could be attributed to the paramagnetic Pd(II) complex.14 Interestingly, five or six sharp peaks appeared when M/L = 1.6 and slowly increased, which is relative to diamagnetic 1:1 complex of ligand and Pd(II). It is hard to give further elucidation of these spectra due to serious peak superposition from the mixture of 1:1 and 1:2 complexes.
 |
| | Fig. 2 (A) Partial enlargement of stacked 1H NMR spectra (500 MHz, 295 K) of 3 (5.3 mM) titrated with Pd(NO3)2 in CD3CN/CDCl3 (7/3, v/v); M/L = Pd equivalents. (B) Color appearance change of 3 (5.3 mM) added with various Pd(II) equivalents in CH3CN/CHCl3 (7/3, v/v); M/L = Pd equivalents. | |
To make the assignment clear, the reverse titration with the ligand adding into the metal solution was then conducted under the identical experimental conditions. In this way, the 1:1 complex is dominantly formed at low L/M values, and in such situation, the peak assignment for 1:1 complex can be undertaken by minimizing the interferences of paramagnetic 1:2 complex.
As seen from the spectra in Fig. 3, upon the ligand addition, a pair of bimodal-peaks relating to Ha, Ha′ and Hb, Hb′, and an unimodal-peak corresponding to Hc, Hc′ in C2 symmetric ligand 3 were all split in pairwise into their counterparts (labeled with  ) in 1:1 complex. This indicates that the C2 symmetry of ligand 3 was broken after the formation of this 1:1 complex with Pd(II). Considering the necessity of four coordination of Pd(II), the structure of this 1:1 complex can be considered as Fig. 4b with one extra nitrate coordinating to Pd(II). Upon more ligand addition, these proton peaks of the ligand 3 are split similarly into five unresolved broad peaks (labeled with
) in 1:1 complex. This indicates that the C2 symmetry of ligand 3 was broken after the formation of this 1:1 complex with Pd(II). Considering the necessity of four coordination of Pd(II), the structure of this 1:1 complex can be considered as Fig. 4b with one extra nitrate coordinating to Pd(II). Upon more ligand addition, these proton peaks of the ligand 3 are split similarly into five unresolved broad peaks (labeled with  ), deriving from the 1:2 complex of ligand with Pd(II). Likewise, the C2 symmetry of ligand 3 in the 1:2 complex shown in Fig. 4c was also broken, but the 1:2 complex might be paramagnetic. Finally, two sets of bimodal-peak companied with one unimodal-peak (labeled with
), deriving from the 1:2 complex of ligand with Pd(II). Likewise, the C2 symmetry of ligand 3 in the 1:2 complex shown in Fig. 4c was also broken, but the 1:2 complex might be paramagnetic. Finally, two sets of bimodal-peak companied with one unimodal-peak (labeled with  ) being slightly overlapped with those of 1:2 complex appeared and grew slowly with further increasing the amounts of 3. This means that a new 1:1 complex (Fig. 4d) with C2 symmetry has been formed in the high ratio of the ligand to nitrate anion. Notably, the twin-peak centered at 3.57 ppm corresponding to Hd, Hd′ was changed in the same fashion with those of Ha to Hc. Thus, three species of the complexes of 3 with Pd(II) were identified by 1H NMR titration in solution and be reasonably assigned as [Pd(3)NO3]+, [Pd(3)2]2+ and [Pd(3)]2+, of which the 1:2 complex is paramagnetic.
) being slightly overlapped with those of 1:2 complex appeared and grew slowly with further increasing the amounts of 3. This means that a new 1:1 complex (Fig. 4d) with C2 symmetry has been formed in the high ratio of the ligand to nitrate anion. Notably, the twin-peak centered at 3.57 ppm corresponding to Hd, Hd′ was changed in the same fashion with those of Ha to Hc. Thus, three species of the complexes of 3 with Pd(II) were identified by 1H NMR titration in solution and be reasonably assigned as [Pd(3)NO3]+, [Pd(3)2]2+ and [Pd(3)]2+, of which the 1:2 complex is paramagnetic.
 |
| | Fig. 3 Stacked 1H NMR spectra (500 MHz, 295 K, 3.55–9.40 ppm) of Pd(NO3)2 (4.5 mM) titrated with various equivalents (0–0.7) of 3 in CD3CN/CDCl3 (7/3, v/v), where L standing for 3,  1:1 [Pd(NO3)(3)]+, 1:1 [Pd(NO3)(3)]+,  1:2 [Pd(3)2]2+, 1:2 [Pd(3)2]2+,  1:1 [Pd(3)]2+. 1:1 [Pd(3)]2+. | |
 |
| | Fig. 4 Structures of ligand 3 and its different complexes with Pd: (a) 3; (b) [Pd(3)(NO3)]+; (c) [Pd(3)2]2+; (d) [Pd(3)]2+. Color codes: C (grey), H (fresh green), N (blue), O (red), Pd (yellow). | |
To further understand the electronic and geometric structure of the above assigned complexes, geometric optimizations using DFT methodology were conducted for [Pd(3)NO3]+, [Pd(3)2]2+ and [Pd(3)]2+. Fig. 4b shows that the optimized structure of 1:1 complex [Pd(3)NO3]+ exhibits an asymmetrical square-planar geometry without coordination of the nitrogen atom N2. Notably, it is perhaps the asymmetrical structure of this complex that results in the three split resonance peaks found in Fig. 3. What's more, the further calculated 1H NMR spectrum for the 1:1 complex [Pd(3)NO3]+ is consistent well with the experimentally titrated spectrum corresponding to this Pd(II) complex. The optimized structure of 1:2 complex [Pd(3)2]2+ gives a C2 symmetry of the whole molecule rather than the ligand itself. It is remarkable that the triplet electronic structure of 1:2 complex is more stable than its singlet by 17.60 kcal mol−1, which confirms that the five broad peaks shown in Fig. 3 are attributed to paramagnetic 1:2 complex. The calculation on 1:1 complex [Pd(3)]2+ found a C2 symmetry structure and a symmetrical square-planar geometry of Pd(II) coordinated with two nitrogen atoms of phenanthroline and two amino nitrogen atoms. Moreover, the calculated 1H NMR spectra for individual optimized geometry shown in Fig. 4 at higher precise basis sets reconfirm the assignment for these experimentally titrated spectra.
ITC studies and binding energies calculations of Pd(II) complexation.
Isothermal titration calorimetry (ITC) test as a sensitive and effective method can give detailed information about thermodynamics processes of complexation reactions and hence the strength of metal–ligand coordination.3d,19 The ITC measurement was carried out in CH3CN solution to determine the association constant (Ka) and thermodynamic parameters of the complexation reactions of Pd(II) with 3. Measured ITC data as shown in Fig. 5 can be fit well with a three step complexation model presented by reactions (1)–(3), where M and L stands for Pd(II) and 3, respectively.| | |
M2+ + L + NO3− = MLNO3+ Ka2
| (2) |
| | |
MLNO3+ + L = ML22+ + NO3− Ka3
| (3) |
 |
| | Fig. 5 Micro-calorimetric titrations of Pd(II) with 3 in CH3CN solution (T = 298 K, I = 0.05 M (Et4)NNO3 in CH3CN). (Up) Raw ITC data for 26 sequential injections (10 μL per injection) of a 3 solution (5.0 mM) into a Pd(II) solution. Initial solution volume: V0 = 1.4 mL, C (Pd(II)) = 1.0 mM. (Down) Net reaction heat collected from the integration of the calorimetric traces. | |
The fitted association constants of Ka1, Ka2 and Ka3 are 6.45 × 103, 7.89 × 103, and 5.42 × 105 respectively as listed in Table 1, indicating that the stability of the three complexes decreases in the order of Pd(NO3)(L) > Pd(L)2 > Pd(L), agreeing with that of 1H NMR titrations roughly. Further computation on the binding energies of the three complexes listing in Table 2 coincided well with the experimental order of their relative stability constants. These fitted results also suggested that the complexation reaction (1) and (2) were driven by enthalpy changes (|ΔH°| > |TΔS°|), while (3) was mainly driven by entropy increase (|ΔH°| < |TΔS°|).20
Table 1 ITC data of the complexation reaction of Pd(II) with ligand 3 measured at 298 K, L = 3, M = Pd
| Complexation reaction |
ΔH° (kJ mol−1) |
TΔS° (kJ mol−1) |
Ka |
| M2+ + L = L2+ |
−23.5 ± 4.59 |
−1.80 |
6.45 × 103 ± 2.60 × 102 |
| M2+ + L + NO3− = MLNO3+ |
−51.8 ± 4.71 |
−19.0 |
5.42 × 105 ± 4.60 × 104 |
| MLNO3+ + L = ML22+ + NO3− |
17.5 ± 1.93 |
39.9 |
7.89 × 103 ± 7.90 × 102 |
Table 2 Binding energies of complexes calculated with DFT method
| Complexes |
Binding energies kcal mol−1 |
Relative binding energies kcal mol−1 |
| [Pd(NO3)(L)]+ |
−570.3 |
0.0 |
| [Pd(L)2]2+ |
−441.3 |
129.0 |
| [Pd(L)]2+ |
−419.7 |
150.6 |
Solvent extraction studies with 19 typical metals
Solvent extraction experiments were then performed to evaluate the capability of 3 to selectively extract Pd(II) over some typical alkali metals, alkaline earth metals, transition metals and typical rare earth elements from HNO3 media. Organic phases of 3 dissolved in octanol (2.0 × 10−3 M) were brought into contact with HNO3 solutions containing 19 typical metals. The distribution ratios (DM) of Pd(II) in comparison with other tested metals at different HNO3 concentrations solutions were shown in Fig. 6. The ligand 3 exhibited high extraction ability for Pd(II) (DPd > 4.0) in the range of 0.4–6.0 M HNO3 solution, while no or weak extraction was observed for other metals (DM < 0.2). The resulting separation factors (SFPd/M) of Pd(II) over the other tested metals including four lanthanide elements were all larger than 20 (Tables S1 and S2 in the ESI†). Based on the similar chemical properties of lanthanide elements, these results demonstrate that 3 is capable of selectively extracting Pd(II) over alkali, alkaline earth, and typical transition metals as well as all the lanthanide metals from highly acidic media.
 |
| | Fig. 6 Influence of HNO3 concentration on the extraction of some typical metals with 3. Organic phase: 2.0 mM 3 dissolved in n-octanol. Aqueous phase: 19 typical metals (0.5 mM each) in HNO3. Contact time: 180 min, T = 298 K, A/O = 1.0, shaking speed: 150 rpm. | |
The results of extraction kinetics studies (Fig. 7) illustrated that the extraction equilibrium for Pd(II) by 3 could be established rapidly and the equilibrium time was merely 5 min, significantly shorter than 15 min and 60 min of current most promising ligands 1 and 2, respectively, for extraction of Am(III) from HLW.5
 |
| | Fig. 7 Influence of contact time on the extraction of some typical metals with 3. Organic phase: 2.0 mM 3 dissolved in n-octanol. Aqueous phase: 19 typical metals (0.5 mM each) in 2.0 M HNO3. T = 298 K, A/O = 1.0, shaking speed: 150 rpm. | |
On the other hand, in an actual solvent extraction separation process, the stripping of Pd(II) from the loaded organic phases was necessary for recovery of Pd(II) and meanwhile recycling the extractant. Therefore, the back-extraction experiments were performed using thiourea with different concentrations in 0.1 M HNO3 solution. As listed in Table 3, the loaded Pd(II) was completely stripped from the organic phases by 10 mM thiourea in 0.1 M HNO3 solution.
Table 3 Stripping of Pd(II) from loaded organic phases (2 mM 3/n-octanol) using thiourea of different concentrations in 0.1 M HNO3 solution. Contact time: 120 min, T = 298 K, A/O = 1.0
| Thiourea concentration (mM) |
1.0 |
2.0 |
4.0 |
5.0 |
10.0 |
| Stripping efficiency (%) |
51.3 |
52.9 |
60.8 |
81.2 |
101 |
As is well known, the phenanthroline- and bipyridine-derived triazine ligands can predominately form 1:2 stable complexes with lanthanide and actinide metals whose typical coordinate number is considered as 9, close to the number of 8 nitrogen atoms providing by two ligands. However, the newly exploited ligand 3 affords mainly the 1:1 complex with Pd(II) in combination with an extra nitride anion, although the less stable 1:2 complex was also observed in 1H NMR titration studies. This may arise from the fundamental difference in the characteristics of central coordination metals together with the structure difference between the side chain of rigid cyclic triazine in ligands 1, 2 and the flexible acyclic side chain in ligand 3.
As shown in Table 4, the computed mean metal–nitrogen bond distances of 2.342 Å in 1:2 complex [Pd(3)2]2+ and 2.285 Å in 1:1 complex [Pd(NO3)(3)]+ are relatively shorter than those of 2.55–2.71 Å in 1:2 complex of 1 and 2 with lanthanide and actinide metals implies the significant difference in the size of recognized cavity formed in each individual complex.
Table 4 Calculated bond distances (Å) of ligand 3 as well as [Pd(NO3)(3)]+, [Pd(3)]2+ and [Pd(3)2]2+ complexes
| Complexes |
M–N1 |
M–N2 |
M–N3 |
M–N4 |
M–O2 |
| [Pd(NO3)(3)]+ |
2.544 |
— |
2.192 |
2.120 |
2.015 |
| [Pd(3)]2+ |
1.983 |
2.185 |
2.173 |
2.173 |
— |
| [Pd(3)2]2+ |
2.473 |
2.185 |
2.367 |
— |
— |
In the optimized geometry of 1:2 complex, a distorted octahedral coordination for Pd(II) with six nitrogen atoms (Fig. 8) must lead to higher tension of bond angles and dihedral angles (see Tables 5 and 6) originating from the coordination of two nitrogen atoms in primary amino groups, thus decreases the stability of the 1:2 complex. However, lower tension of bond angles and dihedral angles in the 1:1 complex [Pd(NO3)(3)]+ adopted typical tetragonal coordination of Pd(II) with 4d8 electronic structure, in combination with a hydrogen bond between a hydrogen atom from amino group and an oxygen atom from the coordinated nitride anion contributes to the notably stability of this complex. In addition, the computation on the minor 1:1 complex [Pd(3)]2+ revealed that the high angle tensions made it the most unstable species among three complexes which is also consistent with its highest binding energies.
 |
| | Fig. 8 The octahedral and distorted octahedral of Pd and six N atoms in 1:2 complex of [Pd(3)2]2+. | |
Table 5 Calculated bond angles (°) the ligand 3 as well as [Pd(NO3)(3)]+, [Pd(3)]2+ and [Pd(3)2]2+ complexes
| Species |
N1–C1–C2 |
N3–C2–C1 |
N3–C2–N5 |
| 3 |
116.13 |
116.28 |
125.87 |
| [Pd(NO3)(3)]+ |
111.45 |
113.88 |
123.85 |
| [Pd(3)]2+ |
112.53 |
122.72 |
122.72 |
| [Pd(3)2]2+ |
116.29 |
119.23 |
123.30 |
Table 6 Calculated torsion angles (°) of the ligand 3 as well as [Pd(NO3)(3)]+, [Pd(3)]2+ and [Pd(3)2]2+ complexes
| Species |
N1–C–C–N2 |
N2–C–C–N3 |
N1–C–C–N4 |
N3–C–N5–N6 |
| 3 |
0.854 |
1.075 |
1.075 |
0.590 |
| [Pd(NO3)(3)]+ |
−1.535 |
−18.487 |
14.419 |
−1.046 |
| [Pd(3)]2+ |
−0.514 |
−10.271 |
−10.271 |
1.529 |
| [Pd(3)2]2+ |
−6.046 |
−22.935 |
−21.521 |
0.812 |
Based on these analyses, a coordination cavity with proper geometry and distances suitable for recognition of Pd(II) was constructed by ligand 3, which may be responsible for the high selectivity towards Pd(II) over other 19 typical metals (SFPd/M > 20). This special extraction selectivity for Pd(II) make the ligand 3 have great advantages compared with the previous soft N-donor ligands8b being reported for separation of Pd(II) from HLLW. Because some other fission products such as Zr(IV), Am(III) could be also co-extracted by these ligands like dicarboxypyridine diamide (DCPDA)21 which is undesired in an actual separation process.8b This is the first time an N-donor phenanthroline-based ligand that has been developed for selective extraction toward Pd from HNO3 solution.
The special difference in extraction rates of ligand 3 for Pd(II) and ligand 1, 2 for Am(III) can be related to the degree of the preorganization of ligands 1, 2, and 3 as shown in Scheme 2. For ligand 2, the more stable trans-conformation must overcome a rotational energy barrier to reach its less-favored cis-conformation for metal binding, which significantly slows down the extraction rate.19b,22 For ligand 1, a conformational change with relative smaller energy barrier from outward–outward or inward–outward to inward–inward configuration is still required prior to metal extraction.19b It is remarkable that the most stable conformation of ligand 3 is completely ready to bind with metal ion, without requiring any conformational change. This results from direct coordination of the unimodal nitrogen atoms to metal in all three complexes identified for ligand 3, rather than the double nitrogen atoms in cyclic triazine in complexes of ligands 1, 2 with Am(III). Therefore, comparing with those of 1:2 complexes [Am(1)2]3+ and [Am(2)2]3+, the more favorable conformation of ligand 3 and entropy change in the reaction of forming 1:1 complex [Pd(NO3)(3)]+ make ligand 3 afford a record extraction rate toward Pd(II).
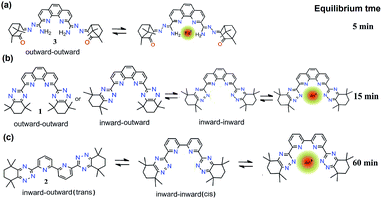 |
| | Scheme 2 The conformation change of the ligands 1, 2 and 3 prior to complexation with metals. | |
Conclusions
In conclusion, we have presented the first example of a promising N-donor phenanthroline-based bis-opened-triazine ligand capable of selective extraction toward Pd(II) with extremely fast rate among 19 typical metals from HNO3 solution. It was demonstrated that the two species of 1:1 [Pd(NO3)(3)]+ and [Pd(3)]2+ complexes were diamagnetic while the distorted octahedral 1:2 complex [Pd(3)2]2+ was paramagnetic. Both the ITC and binding energy calculation studies showed that the asymmetrical 1:1 complex [Pd(NO3)(3)]+ was the most stable of three complexes confirmed in solution, which played a key role in extraction of Pd(II) from HNO3 solution. Further, the preorganization of ligand 3 in a special outward–outward conformation leads to a very high extraction rate for Pd(II). These satisfying extraction performances of ligand 3 towards Pd(II) demonstrated that the selective extraction for some fission products from HLW via accommodating the side chains of phenanthroline skeleton to build a cavity suitable for metal complexation is feasible.
Acknowledgements
The authors would like to acknowledge the financial support from National Natural Science Foundation of China (20871103, 21376210).
Notes and references
- M. Salvatores and G. Palmiotti, Prog. Part. Nucl. Phys., 2011, 66, 144–166 CrossRef CAS.
-
(a) A. Stanculescu, Ann. Nucl. Energy, 2013, 62, 607–612 CrossRef CAS;
(b) Z. Li, P. Cheng, H. Geng, Z. Guo, Y. He, C. Meng, H. Ouyang, S. Pei, B. Sun and J. Sun, Phys. Rev. Spec. Top.--Accel. Beams, 2013, 16, 080101 CrossRef.
-
(a) P. J. Panak and A. Geist, Chem. Rev., 2013, 113, 1199–1236 CrossRef CAS PubMed;
(b) F. W. Lewis, L. M. Harwood, M. J. Hudson, M. G. B. Drew, J. F. Desreux, G. Vidick, N. Bouslimani, G. Modolo, A. Wilden and M. Sypula, J. Am. Chem. Soc., 2011, 133, 13093–13102 CrossRef CAS PubMed;
(c) M. J. Hudson, L. M. Harwood, D. M. Laventine and F. W. Lewis, Inorg. Chem., 2012, 52, 3414–3428 CrossRef PubMed;
(d) Y. Yang, J. Liu, L. Yang, K. Li, H. Zhang, S. Luo and L. Rao, Dalton Trans., 2015, 8959–8970 RSC;
(e) P. K. Mohapatra, A. Sengupta, M. Iqbal, J. Huskens and W. Verboom, Chem.–Eur. J., 2013, 19, 3230–3238 CrossRef CAS PubMed;
(f) C. L. Xiao, C. Z. Wang, L. Y. Yuan, B. Li, H. He, S. Wang, Y. L. Zhao, Z. F. Chai and W. Q. Shi, Inorg. Chem., 2014, 53, 1712–1720 CrossRef CAS PubMed.
-
(a) P. D'Angelo and R. Spezia, Chem.–Eur. J., 2012, 18, 11162–11178 CrossRef PubMed;
(b) ed. J. J. Katz, L. R. Morss, N. M. Edelstein and J. Fuger, The Chemistry of the Actinide and Transactinide Elements, Springer, Netherlands, 2011, vol. 1, pp. 1893–2012 Search PubMed;
(c) The Chemistry of the Actinide and Transactinide Elements, ed. J. J. Katz, L. R. Morss, N. M. Edelstein and J. Fuger, Springer, Dordrecht, 2006, pp. 1–17 Search PubMed;
(d) K. L. Nash, Separation chemistry for lanthanides and trivalent actinides, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner Jr, L. Eyring, G. R. Choppin and G. H. Lander, Elsevier Science, New York, 1994, vol. 18, p. 197–238 Search PubMed.
- A. Geist, C. Hill, G. Modolo, M. Foreman, M. Weigl, K. Gompper and M. J. Hudson, Solvent Extr. Ion Exch., 2006, 24, 463–483 CrossRef CAS.
-
(a) D. Magnusson, B. Christiansen, M. Foreman, A. Geist, J. P. Glatz, R. Malmbeck, G. Modolo, D. Serrano-Purroy and C. Sorel, Solvent Extr. Ion Exch., 2009, 27, 97–106 CrossRef CAS;
(b) E. Aneheim, C. Ekberg, A. Fermvik, M. R. S. J. Foreman, B. Grűner, Z. Hajkova and M. Kvičalová, Solvent Extr. Ion Exch., 2011, 29, 157–175 CrossRef CAS;
(c) E. Aneheim, C. Ekberg and M. R. S. Foreman, Solvent Extr. Ion Exch., 2013, 31, 237–252 CrossRef CAS.
- Z. Kolarik and E. V. Renard, Platinum Met. Rev., 2003, 47, 74–87 CAS.
-
(a) Z. Kolarik and E. V. Renard, Platinum Met. Rev., 2005, 49, 79–90 CrossRef CAS;
(b) R. Ruhela, RSC. Adv., 2014, 4, 24344–24350 RSC.
- C. Madic and M. J. Hudson, Final Report, European
Commission contract no. FI2W-CT91-0112, EUR 18038, 1998 Search PubMed.
- Y. Sasaki, M. Ozawa, T. Kimura and K. Ohashi, Solvent Extr. Ion Exch., 2009, 27, 378–394 CrossRef CAS.
- M. Yu. Alyapyshev, V. A. Babain, L. I. Tkachenko, I. I. Didenko, A. V. Eliseev and M. L. Petrov, Solvent Extr. Ion Exch., 2011, 29, 619–636 CrossRef CAS.
- C. Ekberg, E. Löfström-Engdahl, E. Aneheim, M. R. Foreman, A. Geist, D. Lundberg, M. Denecke and I. Persson, Dalton Trans., 2015, 18395–18402 RSC.
- Ž. D. Bugarčić, J. Bogojeski and R. v. Eldik, Coord. Chem. Rev., 2015, 292, 91–106 CrossRef.
- K. R. Dunbar and J. S. Sun, J. Chem. Soc., Chem. Commun., 1994, 20, 2387–2388 RSC.
- S. F. Boys and F. Bernardi, Mol. Phys., 2006, 19, 553–566 CrossRef.
- S. Simon, M. Duran and J. J. Dannenberg, J. Chem. Phys., 1996, 105, 11024–11031 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. Afsar, D. M. Laventine, L. M. Harwood, M. J. Hudson and A. Geist, Chem. Commun., 2013, 49, 8534–8536 RSC.
-
(a) M. Ostermeier, M. A. Berlin, R. M. Meudtner, S. Demeshko, F. Meyer, C. Limberg and S. Hecht, Chem.–Eur. J., 2010, 16, 10202–10213 CrossRef CAS PubMed;
(b) F. W. Lewis, L. M. Harwood, M. J. Hudson, M. G. Drew, V. r. Hubscher-Bruder, V. Videva, F. O. Arnaud-Neu, K. Stamberg and S. Vyas, Inorg. Chem., 2013, 52, 4993–5005 CrossRef CAS PubMed.
-
(a) S. O. Kang, V. M. Lynch, V. W. Day and E. V. Anslyn, Organometallics, 2011, 30, 6233–6240 CrossRef CAS PubMed;
(b) J. J. Henkelis and M. J. Hardie, CrystEngComm, 2014, 16, 8138–8146 RSC.
- M. Y. Alyapyshev, V. A. Babain, Y. A. Pokhitonov and V. M. Esimantovskiy, Proc. Conf. (Global 2007), 9–13 Sep 2007, Boise-Idaho, United States, p. 1873 Search PubMed.
- M. R. S. Foreman, M. J. Hudson, M. G. B. Drew, C. Hill and C. Madic, Dalton Trans., 2006, 1645–1653 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra20484h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 





 ) in 1
) in 1 ), deriving from the 1
), deriving from the 1 ) being slightly overlapped with those of 1
) being slightly overlapped with those of 1
 1
1 1
1 1
1




