
Synthesis and characterization of donor–acceptor type conducting polymers containing benzotriazole acceptor and benzodithiophene donor or s-indacenodithiophene donor
Di Zhangab,
Min Wangc,
Xiaoli Liua and
Jinsheng Zhao*a
aShandong Key Laboratory of Chemical Energy Storage and Novel Cell Technology, Liaocheng University, Liaocheng, 252059, P. R. China. E-mail: j.s.zhao@163.com; Fax: +86-635-8539607; Tel: +86-635-8539607
bCollege of Chemical Engineering, China University of Petroleum (East China), QingDao, 266580, P. R. China
cLiaocheng People's Hospital, Liaocheng, 252000, P.R.China
First published on 20th September 2016
Abstract
Two novel electrochromic copolymers, poly(benzodithiophene-benzotriazole) (PBDBT) and poly(s-indacenodithiophene-benzotriazole) (PIDBT) were synthesized successfully through Stille coupling reactions. Both polymers were characterized by cyclic voltammetry (CV), UV-Vis spectroscopy, density functional theory (DFT), colorimetry and thermal gravimetric analysis. The band gaps of the two polymers calculated according to spectroelectrochemistry data are 1.90 eV for PBDBT and 1.93 eV for PIDBT, respectively. The PBDBT film turns rosybrown to transparent grey when it is oxidized and the PIDBT film turns pale violet-red in the neutral state to transparent light grey in the oxidized state. PBDBT has an 55.0% optical contrast at 1500 nm, PIDBT has an 60.8% optical contrast at 1560 nm, and their corresponding response times are 1.49 s and 1.85 s, respectively. In addition, the coloration efficiency of PBDBT at 1500 nm is 144.70 cm2 C−1, and the coloration efficiency of PIDBT at 1560 nm is 222.00 cm2 C−1. Both copolymers demonstrate good properties, and can be promising electrochromic materials for commercial applications.
Introduction
Conducting polymers have received more and more attention due to their wide range of applications, such as in organic light emitting diodes (LEDs),1,2 lasers,2 image sensors3 and display applications.4 The advantages manifested by them consist of high optical contrast, fast response time, flexible processability and fine tuning.5 Electrochromic materials can display at least two different colours when appropriate current density is applied to the electrode,4 and among them, polythiophene and its derivatives have been widely researched.6,7 Polarons and bipolarons are two kinds of charge carrier in conducting polymers, which is confirmed by theories and experiments.8,9The band gap values are regarded as the vital parameters resolving the electrochromic characteristics of polymers,2,10 which could be tuned by some feasible strategies including bond-length alternation, inducing order, creating an planar structure and adopting donor–acceptor (D–A) approach.7 D–A approach is the most common and useful method among them. It functions by the push–pull interaction in the molecular backbone, which can facilitate intramolecular charge transfer.11 The electron donor group can raise highest occupied molecular orbital (HOMO) energy, making oxidation easy to achieve, and the acceptor group can lower the lowest unoccupied molecular orbital (LUMO) energy, which can promote the reduction process.5,12 Vinylene, thiophene, 3,4-ethylenedioxythiophene (EDOT) are electron-rich units, while quinoxaline, 2,1,3-benzothiadiazole, 2,1,3-benzoselenadiazole, thienopyrazine13 are usually electron-deficient units.
Taking benzotriazole as acceptor units, some D–A type polymers have been prepared and be proven to be promising candidates as electrochromic or solar energy generation materials, although the electron affinity ability of benzotriazole is weaker than that of the majority of electron withdrawing units mentioned above.14 Abidin Balan and his colleagues synthesized one polymer taking EDOT as donor unit and benzotriazole as acceptor unit, and the resulting copolymer showed superior properties in many aspects.15 When changing the donor unit from EDOT to thiophene, Abidin Balan synthesized another copolymer monomer, 2-dodecyl-4,7-di(thiophen-2-yl)-2H-benzo[d][1,2,3]triazole (TBT), which could be polymerized to a polymer by the electrodeposition method, the obtained polymer could display three primary colours, changing from red to green and final blue when it is oxidized, while when it is n-doped, its colour could change to light blue, and eventually transparent.16 When the alkyl group is substituted on the thiophene unit, the steric repulsion effects could be anticipated between the repeating units along the backbone of the polymer chains, which could be avoided while placing the alkyl group on the nitrogen atom of benzotriazole unit, and then making the resultant copolymer more planar.17
Benzodithiophene has a high hole mobility and can form π–π stacking easily due to its large planar structure,13,18 which was proved by the X-ray diffraction (XRD) analysis.18 Jianhui Hou and his colleagues investigated the copolymers based on benzodithiophene in details, and their band gaps can be tuned between 1.0 eV to 2.49 eV by different combined manner.19 Although the steric hindrance could affect the band gap, in case of the benzodithiophene unit, the alkoxyl chains exist on the side positions including 4 and 9 positions, which reduce the steric hindrance effects to a very low degree.19 So, benzodithiophene and its derivatives are most popular donor units for the fabrication of D–A type conjugated polymers.20 When benzodithiophene unit is taken as the donor core, the introduction of the alternate electron deficient units in the polymer chain could generate intra-molecular charge transfer, and consequently the copolymer has a small bad gap.21
Polymers incorporating 4,9-dihydro-s-indaceno[1,2-b:5,6-b′]dithiophene (IDT) as the donor units typically have excellent solubilities.22 Planarity plays an important role in molecular packing, and influencing not only the conjugation length but also the charge transfer speeds within the polymer films stacked by individual polymer molecules.23 As known to us, IDT possesses ladder planar structure, which can extend the π electron delocalization of the polymer backbone containing it, making it a good electron donor.22 Taking IDT as the donor unit, D–A type conjugated polymer could be achieved through a copolymerization reaction with appropriate acceptor unit, and the resultant polymer has been applied to field effect transistors24 and solar cells.23 Wei Teng Neo and his colleagues firstly applied IDT-containing polymers to electrochromic area.22 The reports about copolymers containing the above mentioned units for electrochromic applications are still very limited, further efforts in this area should be strengthened.
In this study, two D–A type conducting polymers, poly(benzodithiophene-benzotriazole) (PBDBT) and poly(s-indacenodithiophene-benzotriazole) (PIDBT) were synthesized by Stille coupling reaction, and then spray casting method was used to pattern a copolymer film on ITO conducting glass. Finally, a series of characterization methods, including electrochemistry, spectroelectrochemistry, density functional theory (DFT) calculation, colorimetry, kinetics and thermogravimetry, were carried out for the detailed acquaintance of polymers, and the results witnessed the excellent electrochromic properties of the polymers. In this case, both of the polymers could be potential candidates for the fabrication of electrochromic device.
Experimental
Materials
Bis-(triphenylphosphine)palladium(II) chloride (Pd(PPh3)2Cl2, 99%), N-bromosuccinimide (NBS, 99.0%), acetonitrile (ACN), tetra-n-butylammonium hexafluorophosphate (TBAPF6, 98%) were all purchased from Aladdin Co., Ltd and used without further purification. Chloroform (99.0%), acetone (99.5%), methanol (99.5%), ethanol (95%) and other solvents were used directly when received from Laiyang Fine Chemical Factory. Sodium sulfite (Na2SO3), magnesium sulfate (MgSO4, ≥99.0%) and other reagents were bought from Tianjin Chemreagent Co., Ltd. and used directly. (4,8-Bis((2-ethylhexyl)oxy)benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl)bis(trimethylstannane) (BT) (99%), (4,4,9,9-tetrakis(4-hexylphenyl)-4,9-dihydro-s-indaceno[1,2-b:5,6-b′]dithiophene-2,7-diyl)bis(trimethylstannane) (IDT) (99%) were purchased from Zhengzhou Alfachem Co., Ltd (Zhengzhou, China). Indium-tin-oxide (ITO) coated glass (sheet resistance: < 10 Ω □−1, purchased from Zhuhai kaivo Co., Ltd) were washed with ethanol before use. The electrolyte in the experiments was 0.1 M TBAPF6/ACN.Instrumentation
1H NMR and 13C NMR spectra of the products were recorded with a Varian AMX 400 spectrometer. The electrochemical characteristics were fulfilled by cyclic voltammetry (CV) using a Ivium-n-Stat Analyzer, where ITO glass, a Pt and an Ag wire were employed as working, counter and pseudo reference electrode, respectively. Spectroelectrochemical properties were measured with a Varian Cary 5000 spectrophotometer, and the potential of the polymer film (spray coated on ITO glass) to be tested was controlled by the above electrochemical analyzer synchronously. Colorimetry was also investigated through the Varian Cary 5000 spectrophotometer. Digital images of the polymer were taken with a common digital camera. The polymer films were sprayed onto ITO conducting glasses with airbrush after they were dissolved in chloroform (5 mg mL−1).Synthesis of 4,7-bis(5-bromothiophen-2-yl)-2-dodecyl-2H-benzo[d][1,2,3]triazole
The precursors of 2-dodecyl-4,7-di(thiophen-2-yl)-2H-benzo[d][1,2,3]triazole (TBT) were synthesized as the method of literature.25 Then 4,7-bis(5-bromothiophen-2-yl)-2-dodecyl-2H-benzo[d][1,2,3]triazole was synthesized by the bromination of NBS as the previous literature25,26 and showed in Scheme 1. In a flask, 2.0 g of TBT (0.030 mol L−1) and 1.89 g of NBS (0.071 mol L−1, 2.4 equiv.) was dissolved in a mixture of 150 mL of CH2Cl2/glac CH3COOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solvent. The mixture was refluxed with stirring for 24 hours in the dark and then poured into 200 mL of deionized water. The organic phase was separated and poured into 100 mL of saturated Na2SO3. After 4 times of extraction, the solvent in organic phase was distilled and column chromatograph was finished with the eluent (DCM: n-hexane). Finally, we can get yellow crystallized solid product. 1H NMR (400 MHz, CDCl3, δ ppm) 7.78 (d, 2H), 7.48 (s, 2H), 7.12 (d, 2H), 4.78 (t, 2H), 2.17 (m, 2H), 1.48–1.15 (m, 18H), 0.88 (t, 3H); 13C NMR (101 MHz, CDCl3, δ ppm) 141.62, 141.15, 130.81, 126.88, 122.89, 122.11, 113.10, 56.87, 31.87, 29.99, 29.58, 29.51, 29.40, 29.30, 28.96, 26.52, 22.65, 14.10.
:1) solvent. The mixture was refluxed with stirring for 24 hours in the dark and then poured into 200 mL of deionized water. The organic phase was separated and poured into 100 mL of saturated Na2SO3. After 4 times of extraction, the solvent in organic phase was distilled and column chromatograph was finished with the eluent (DCM: n-hexane). Finally, we can get yellow crystallized solid product. 1H NMR (400 MHz, CDCl3, δ ppm) 7.78 (d, 2H), 7.48 (s, 2H), 7.12 (d, 2H), 4.78 (t, 2H), 2.17 (m, 2H), 1.48–1.15 (m, 18H), 0.88 (t, 3H); 13C NMR (101 MHz, CDCl3, δ ppm) 141.62, 141.15, 130.81, 126.88, 122.89, 122.11, 113.10, 56.87, 31.87, 29.99, 29.58, 29.51, 29.40, 29.30, 28.96, 26.52, 22.65, 14.10.
 | ||
| Scheme 1 Synthetic routes of monomers and copolymers PBDBT and PIDBT. | ||
Synthesis of PBDBT
PBDBT and PIDBT were copolymerized as displayed in Scheme 1. 0.6245 g of DBTBT, 0.7723 g of BT (1 mmol), 0.02804 g of Pd(PPh3)2Cl2 (0.04 equiv.) and 100 mL of toluene were mixed in a flask and refluxed for 48 hours in argon atmosphere.25 After the toluene was distilled off, the crude product was dissolved in a small amount of chloroform, and the solution was added dropwise into 100 mL of cold methanol, solid powder (crude polymer) was precipitated on the bottom of the flask, and collected by filtration. The resulting solid powder was extracted in the Soxhlet extractor with methanol and acetone as the solvent, successively for 24 hours, until each of the solvent was transparent. 1H NMR (400 MHz, CDCl3): 7.99 (d, 2H), 7.76 (s, 2H), 7.69 (s, 2H), 7.33 (d, 2H), 4.81 (t, 2H), 4.35 (d, 4H), 2.19 (m, 2H), 0.88–1.85 (m, 51H).Synthesis of PIDBT
0.3747 g of DBTBT (0.6 mmol), 0.7398 g of IDT (0.6 mmol, 1 equiv.) and 0.01685 g of Pd(PPh3)2Cl2 (0.04 equiv.) were dissolved in 100 mL of toluene in 250 mL of three-necked flask, and then the mixtures were refluxed for 48 hours under argon atmosphere. When the reaction was finished, it was treated as the method of PBDBT. 1H NMR (400 MHz, CDCl3): δ 8.00 (s, 2H), 7.57 (s, 4H), 7.41 (m, 4H), 7.24–7.03 (m, 16H), 4.83 (s, 2H), 2.58 (d, 8H), 2.20 (s, 2H), 1.54–1.01 (m, 50H), 0.87 (t, 15H).Results and discussion
Electrochemistry
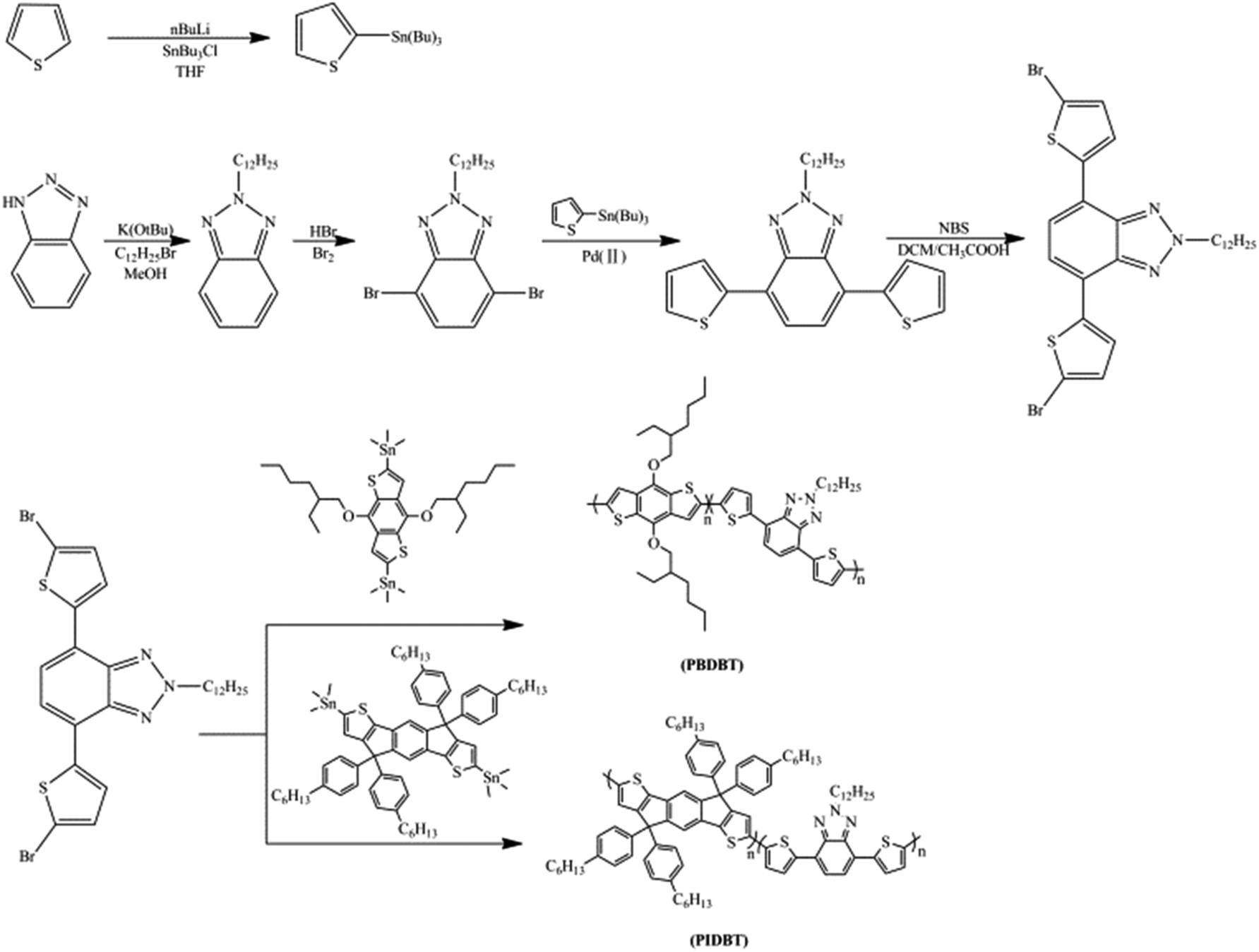
CVs were conducted for the determination of the redox properties of the polymers. The solid copolymers PBDBT and PIDBT were dissolved in CHCl3, then spray coated on the ITO glasses until the solvent was evaporated thoroughly. As a result, some copolymer thin films coated on the glasses were achieved and placed in 0.1 M TBAPF6/ACN solution. As shown in Fig. 1, two groups of redox peaks occurred in the positions of 1.08 V/1.28 V and −1.43 V/−1.35 V for PBDBT when the potentials were cycled from −2.0 V to 2.0 V. Similarly, the redox peaks located at 1.61 V/0.87 V and −1.56 V/−1.25 V for PIDBT, respectively, when the potentials were cycled between −2.0 V to 2.5 V.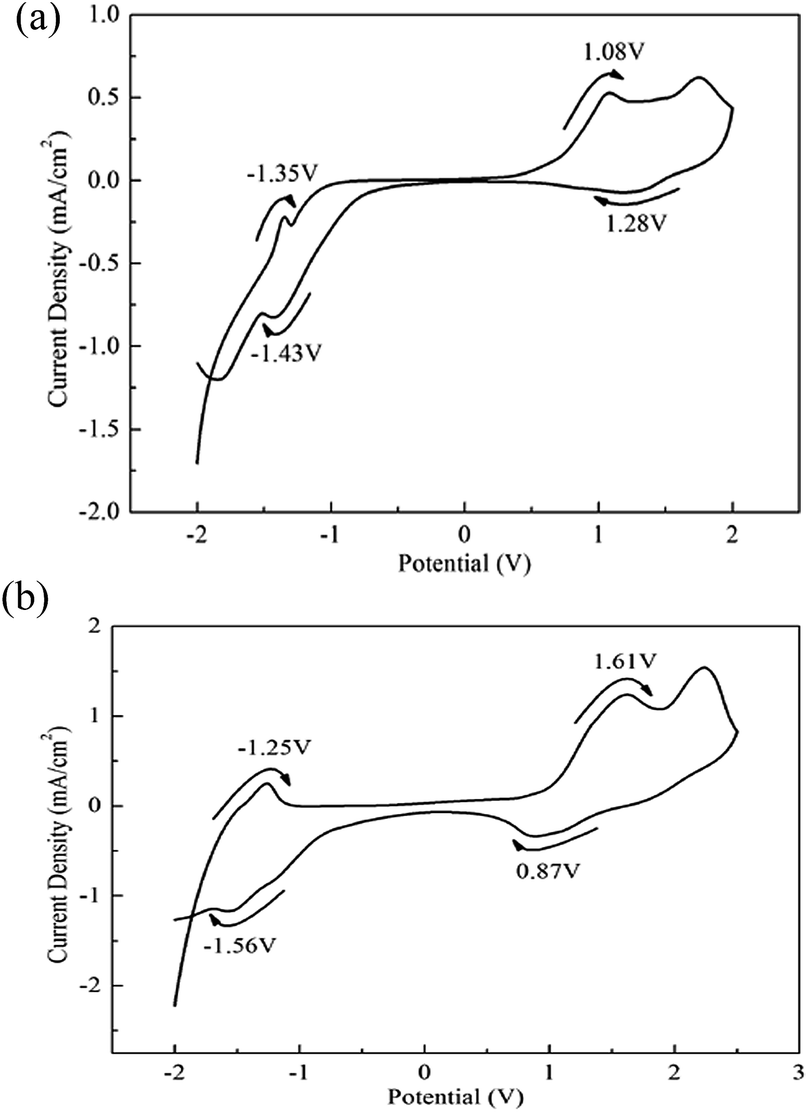 | ||
| Fig. 1 CV of spray coated PBDBT (a) film from −2.0 V to 2.0 V and PIDBT (b) film from −2.0 V to 2.5 V. | ||
The initial redox peaks at the positive potentials suggested the existence of the p-type doping, which might be related with the electron-donating property of benzodithiophene or indacenodithiophene.26 In D–A type polymer, the HOMO values of the donor unit is approximately equal to the values of the polymer, which is then determined by the initial oxidative potential (Eonset) of the polymer. The stronger electron donating ability of the donor unit is beneficial to reduce the Eonset of the polymer, and finally lead to the reduced Eg of the polymer. With the potential increased, copolymers lose electrons, the first peak appearing. The Eonset value of PBDBT (0.64 V) is lower than that of PIDBT (1.01 V), demonstrating the stronger electron-donating ability of benzodithiophene than that of indacenodithiophene unit (Fig. 1). And, when the potentials decrease from the positive potentials to 0 V, the polymers restored to their neutral states. Both copolymers have n-type doping abilities and the initial negative potentials denote n-type doping. The n-type doping voltage of PBDBT is larger (−1.43 V) than PIDBT (−1.56 V), signifying the PBDBT has the stronger electron-withdrawing ability (Fig. 1).25
Optical properties of the copolymer solution and films
As shown in Fig. 2, the UV-Vis absorption spectra of PBDBT in solution was obtained through dissolving the product in CHCl3, and that of PBDBT film was obtained through placing the polymer coated ITO electrode in ACN solution. The occurrence of absorption peaks could be ascribed to the π–π* conjugation system.27 The absorption peaks of the copolymers in solution are located at 532 nm and 531 nm for PBDBT and PIDBT, respectively, nearly located in the same position, except that the width of the absorption band of the polymer PBDBT is somewhat wider than that of the polymer PIDBT. The reason for the difference in the width of the absorption band may be that the molecular weight distribution of the polymer PBDBT is wider than that of the polymer PIDBT. They displayed two different colours, medium violet red for PBDBT, and magenta for PIDBT.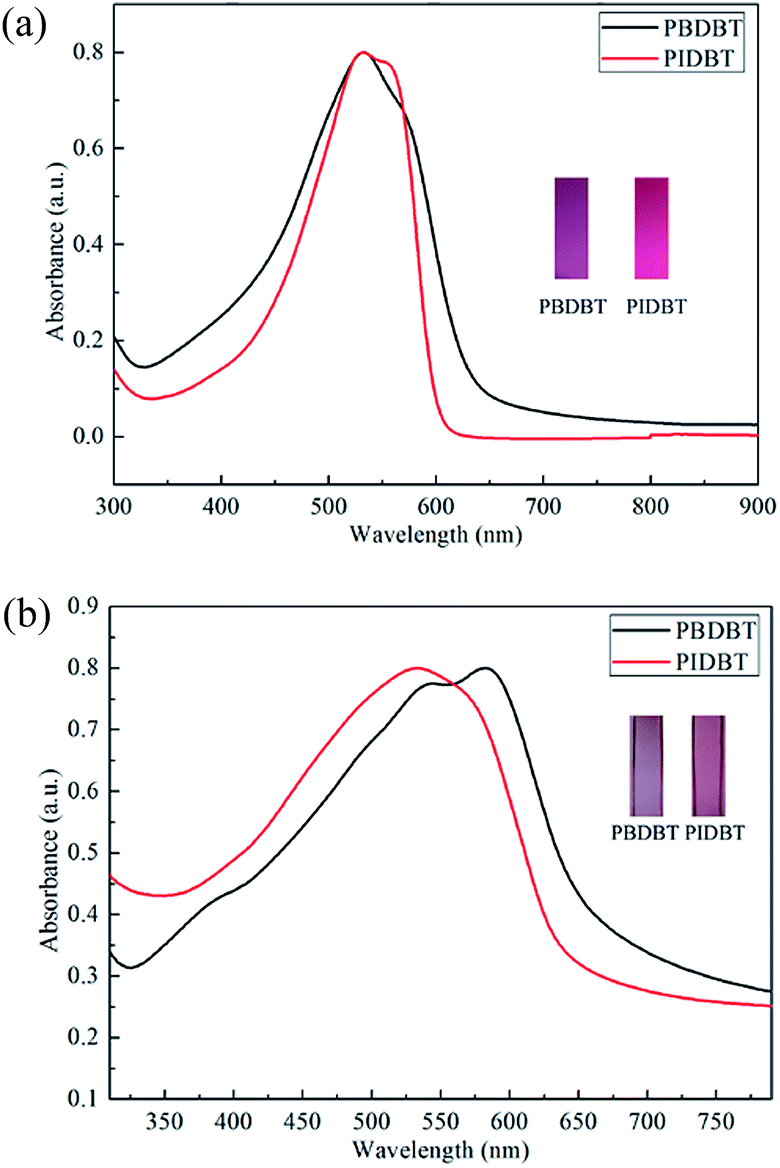 | ||
| Fig. 2 UV-Vis absorption spectra of PBDBT and PIDBT dissolved in CHCl3 (a) and those of their films coated on ITO electrode in TBAPF6/ACN (b) in neutral state. | ||
The absorption peaks located at 532 nm in solution and 582 nm in film solid state for PBDBT was observed, and 531 nm in solution and 536 nm in film state were also found for PIDBT, respectively. Compared with the absorption in the solution, there is an apparent red shift for the polymers existing in the form of film, which is ascribed to the better interchain interaction stemmed from π–π stacking of the copolymers.28 The colour of PBDBT film and PIDBT film are rosybrown and pale violet red, respectively, at the neutral state. There is an obvious bathochromic shift for PBDBT films (582 nm) compared with PIDBT films (536 nm) in neutral state, manifesting that PBDBT has a smaller energy absorbance. In other words, PBDBT need a small energy to run π–π* transition and it has a more conjugated structure, which is anticipated by researchers.
Morphology analysis
Atomic force microscopy (AFM) was used for the morphology analysis of the polymers. As shown in Fig. 3, both polymer films presented homogenous and smooth morphology with the mean roughness (Ra) of 18.3 nm and 27.9 nm, respectively, for PBDBT and PIDBT. Although no direct evidence has been witnessed that there is a definite relationships between the morphologies of polymer films and their electrochromic properties, the smooth morphologies of two polymers at least suggested their perfect film-processing capabilities, which is conducive to the preparation of large area electrochromic device and even for commercial applications. | ||
| Fig. 3 AFM image of PBDBT (a) and PIDBT (b). | ||
Spectroelectrochemistry
Spectroelectrochemistry is an effective and widely applied method to appraise the electrochromic properties of conducting polymers. When oxidation potential rises gradually, the apparent colour changes could be observed. Fig. 4 displays the spectroelectrochemistry property of PBDBT and PIDBT under stepwise oxidation, as well as the photos in neutral states and oxidized states. In neutral states, there is only one absorption band for each polymer due to π–π* transition in the visible region. When the voltage is applied to the copolymer films, the charge carriers appear, including polarons and bipolarons. | ||
| Fig. 4 Spectroelectrochemistry of PBDBT (a) and PIDBT (b) and their photos in neutral states and oxidized states. | ||
For PBDBT, with potential increasing, the initial peak located at 578 nm decreases, while another two new peaks appear in near infrared region at 781 nm and 1388 nm, respectively. The peak at 781 nm is because of the formation of polaron (singly charged species), while the peak at 1388 nm which located in a low energy region can be ascribed to the bipolaron.29 The polaron peak reaches largest when the potential is up to 1.42 V, while the bipolaron peak is rising with the increase of voltage, which is assigned to the repression of inter-band transition.28,30 Same with PBDBT, the three peaks of PIDBT are at 532 nm, 841 nm, 1390 nm corresponding to π–π* transition, formation of polaron and bipolaron, respectively. The polaron peak reaches a maximum when the applied potential is 1.22 V, and the bipolaron peak is increasing when the potential is turning large.
As shown in Fig. 4(a), with the applied potential increasing, the colours of PBDBT film change from rosybrown in neutral states to transparent grey in oxidized states. White light is a mixture of light that is formed by the superposition of various wavelengths of light. When the potential changes, the absorption of light of different frequencies varied, and generated different colours. In neutral states, there is an absorption peak located mainly in green light region, so the film demonstrates rosybrown. With the potential increasing, the absorption of green light deceases, while the absorption of red light increases. Finally, the absorption of two peaks in visible region is nearly equal, therefore, the film is transparent grey in oxidized states. Similarly, PIDBT also experiences an palpable colour change from pale violet red to transparent light grey after it is oxidized.
Some parameters of spectroelectrochemistry of PBDBT and PIDBT were read from their graphs or calculated according to previous studies27,31 and were summarized in Table 1, including onset of the optical absorption spectra in neutral states (λonset), maximum absorption wavelength of PBDBT and PIDBT in solution (λmax, solution) and film (λmax, film) morphology, onset oxidation potential (Eonset), optical band gap (Eg) and HOMO/LUMO energy levels of both copolymers. Eg, EHOMO and EHOMO were calculated with the formula Eg = 1241/λonset, EHOMO = −e(Eonset + 4.4) and ELUMO = EHOMO + Eg, respectively.27,31
The Eg values of the two polymers are 1.90 eV and 1.93 eV, respectively, and the HOMO values exist a large diversity. The electron donor group can raise HOMO energy, while the acceptor group can lower LUMO energy.5,12 Since both polymers have same acceptor unit, the difference of HOMO can be assigned to the donor unit. In case of benzodithiophene unit, benzene links with two thiophene directly, while in case of s-indacenodithiophene unit, benzene is connected with thiophene through cyclopentene, so benzodithiophene has a better planarity and more conjugated structure than s-indacenodithiophene. Consequently, PBDBT has a smaller band gap and a larger HOMO value than that of PIDBT. As a kind of homopolymer of benzodithiophenes, poly(4,8-bis(dodecyloxy)benzo[1,2-b:4,5-b′]dithiophene) (H2, shown in Table 1) has a band gap of 2.13 eV,19 which was higher than that of PBDBT (1.90 eV). The introduction of TBT unit is beneficial to lower the band gap of the resultant copolymers, and also demonstrates the advantages of donor–acceptor approach for this purpose. Compared with H2, the maximum absorption wavelength of PBDBT has a red shift no matter for polymer solution or polymer film, which could be ascribed to the better conjugated structure of PBDBT backbone than that of H2.
Similarly, a recent reported copolymer of substituted s-indacenodithiophene poly(2-([2,2′-bithiophen]-5-yl)-4,4,9,9-tetrakis(4-(octyloxy)phenyl)-4,9-dihydro-s-indaceno[1,2-b:5,6-b′]dithiophene) (P3, Table 1), has a slightly higher band gap than that of PIDBT due to the introduction of the bithiophene and other than the TBT unit into the copolymer.22 Because benzo[d][1,2,3]triazole is a good acceptor unit, when it is introduced to the polymer backbone, the push–pull interaction will promote the carrier transfer and enhance the delocalized structure, and then lower the band gaps of the copolymers.11 More specifically, the introduction of the benzo[d][1,2,3]triazole unit has a pronounced impact on the LUMO energies of the copolymers, as a resultant, the LUMO value of PIDBT is much lower than that of P3.
Density functional theory calculation
The frontier molecular orbital of the two monomers were calculated by density functional theory (DFT) using Gaussian 09 software at the parameter of 3-21G basic set. The charge distribution is shown in Fig. 5. The values of HOMO and LUMO are −5.20 eV, −2.53 eV for BDBT and −4.85 eV, −2.43 eV for IDBT, respectively, and the theoretical band gaps of both monomers are 2.66 eV for BDBT and 2.42 eV, for IDBT, respectively. The band gap of BDBT is larger than IDBT, this tendency is inconsistent with their copolymers in optical experiments. This phenomenon may be explained by that BDBT monomer is smaller than IDBT, and the conjugated scale is not as large as later, as a result, the delocalization of molecule is not so sufficient as IDBT. In addition, in DFT calculation, we neglect the effect of solvent, and in the experiments, copolymers existed in solid film, which exists π–π stacking.32,33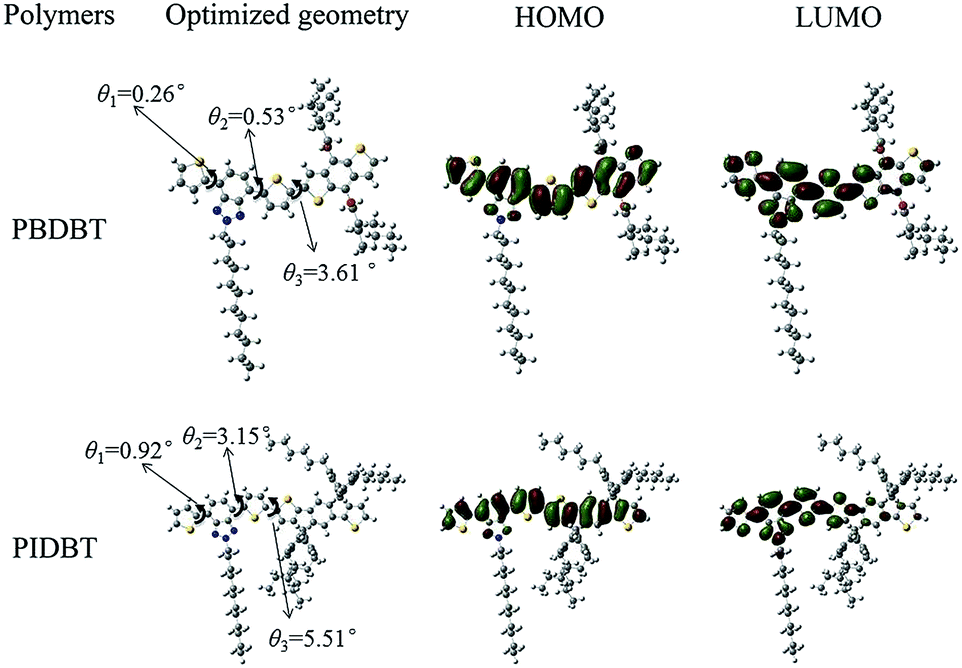 | ||
| Fig. 5 Molecular orbital diagrams of HOMO and LUMO of PBDBT and PIDBT monomers. | ||
In addition, we should notice that the dihedral angles in both monomers have an obvious diversity. θ1 and θ2 are the dihedral angles in benzotriazole unit, and θ3 is the dihedral angle between benzotriazole unit and benzodithiophene unit or s-indacenodithiophene unit. θ1, θ2, θ3 are 0.26°, 0.53°, 3.61° for BDBT, and 0.92°, 3.15°, 5.51° for IDBT. The dihedral angle of BDBT monomer is smaller than IDIT, indicating that BDBT had a more obvious planar structure. Since planar structure can extend π electron delocalization, which is beneficial to reducing the band gap, so the band gap of PBDBT is smaller than PIDBT, which is consistent with the experiment.
Electrochromic switching of PBDBT and PIDBT
It's important for conducting polymers to have a fast switching property and to change colour quickly when the copolymer films change from neutral state to oxidized state. Electrochromic switching properties were studied through observing the change of transmittance with time. Square wave potential with an interval of 4 s was applied into the film when multi-potential steps technique was chosen and parameters were all set up in CHI 760 C.32,34 The potential were interchanged between 0 V and 1.45 V for PBDBT and −0.3 V and 1.4 V for PIDBT. Fig. 5 demonstrates the change of transmittance with time.The optical contrast (ΔT%) is the change in transmission rate of light between the neutral and doped state, which is important to evaluate the property of polymers.32 As shown in Fig. 6, the ΔT% of PBDBT are 16.8% at 580 nm, 15.7% at 850 nm and 55.0% at 1500 nm, and the ΔT% of PIDBT are 32.0% at 570 nm, 26.2% at 850 nm and 60.8% at 1560 nm. Both polymers had large ΔT%, demonstrating that the colour change is obvious. Besides, the range of transmittance in near infrared region is larger than that in visible region, indicating good potential NIR applications.
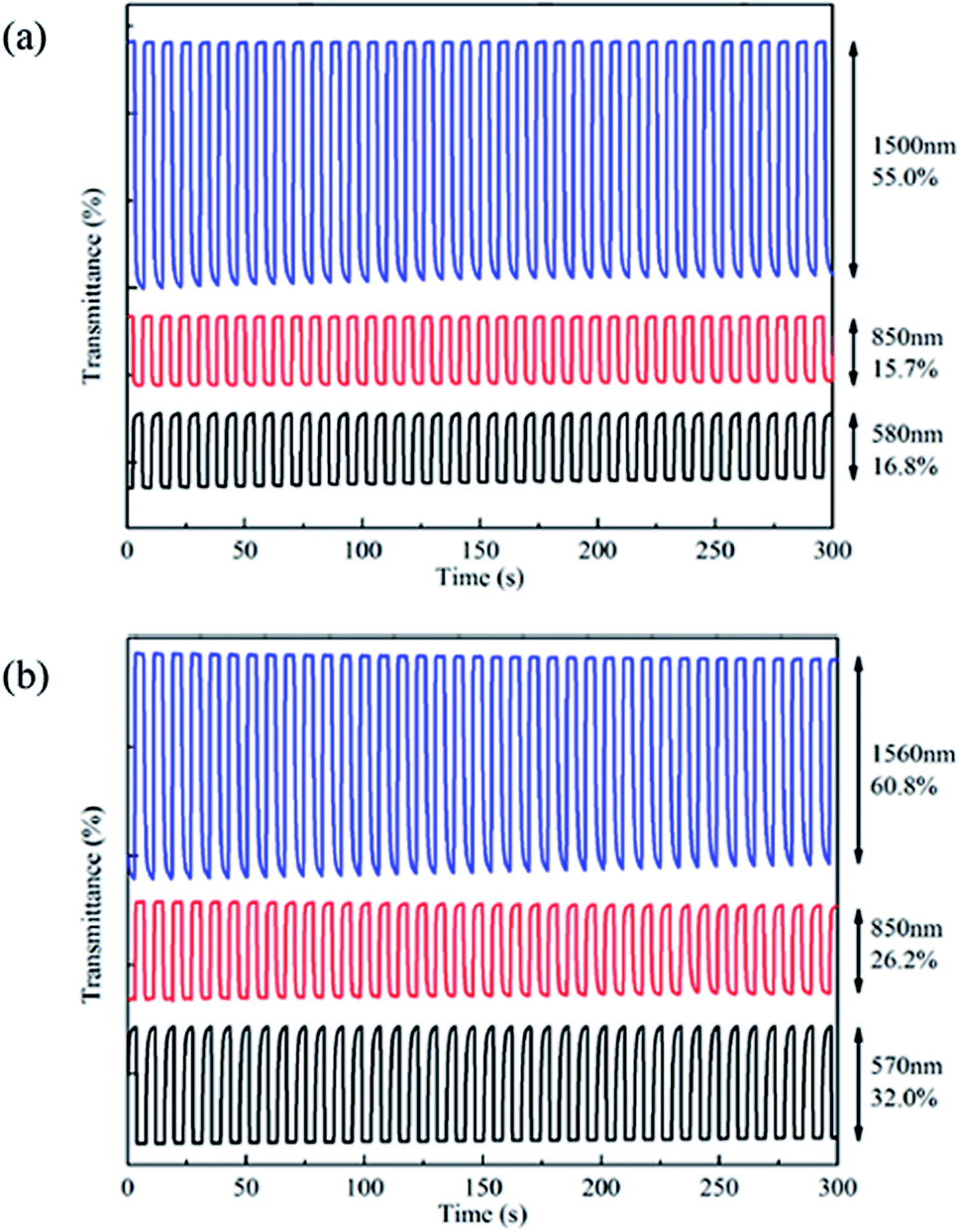 | ||
| Fig. 6 Electrochromic switching of PBDBT (a) between 0 V and 1.45 V and PIDBT (b) between −0.3 V and 1.4 V. | ||
Response time (t95%), reflecting the speed switching between neutral state and oxidized state, is defined as the time needed for obtaining 95% of the entire optical contrast, because the naked eye isn't very sensitive to the remaining 5% changes.35 As shown in Table 2, the response time of PBDBT are 1.01 s at 580 nm, 0.75 s at 850 nm and 1.49 s at 1500 nm, and the response time of PIDBT are 1.84 s at 570 nm, 0.66 s at 850 nm and 1.85 s at 1560 nm, respectively. Both copolymers exhibited outgoing switching times.
| Copolymers | λ, nm | ΔT%, % | Response time (t95%), s | Coloration efficiency (η), cm2 C−1 |
|---|---|---|---|---|
| PBDBT | 580 | 16.8 | 1.01 | 69.16 |
| 850 | 15.7 | 0.75 | 66.93 | |
| 1500 | 55.0 | 1.49 | 144.70 | |
| PIDBT | 570 | 32.0 | 1.84 | 143.22 |
| 850 | 26.2 | 0.66 | 85.75 | |
| 1560 | 60.8 | 1.85 | 222.00 |
Coloration efficiency (η) is taken for evaluating the utilization efficiency of the electronic power during switching, and defined as the ratio of optical density change (ΔOD) and electric charge per unit area (ΔQ) and illustrated by the following equations.32
| η = ΔOD/ΔQ |
| ΔOD = lg(Tb/Tc) |
| ΔQ = Q/A |
Changing the interval in multi-potential steps from 10 s to 4 s, 2 s, and finally to 1 s, we can compare the decline degree of optical contrast. As shown in Fig. 7, when the interval decreases from 10 s to 1 s, PBDBT maintains an optical contrast of 16.5% at 580 nm with a 2.6% decrease, an optical contrast of 15.5% at 850 nm with only a 1.1% drop and an optical contrast of 45.4% at 1500 nm with a 8.2% decrease, while PIDBT maintains an optical contrast of 24.0% at 570 nm with a 12.6% decrease, an optical contrast of 21.7% at 850 nm with a 6.0% drop and an optical contrast of 47.2% at 1560 nm with a 14.7% decrease. Compared with PIDBT, PBDBT decreases inconspicuously, which may be ascribed to its good planarity, promoting the transfer speed of electron.
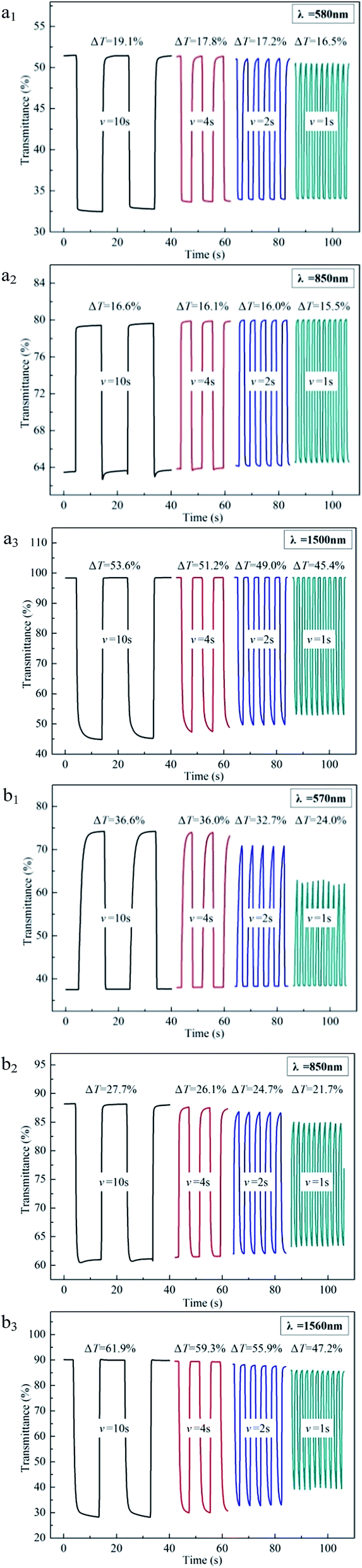 | ||
| Fig. 7 Electrochromic switching of PBDBT (a1–a3) and PIDBT (b1–b3) with an interval of 10 s, 4 s, 2 s and 1 s. | ||
Colorimetry
Colorimetry is one important method to evaluate the change of colour. In this study, CIE 1976 L*a*b* colour space was used to measure the colour at different potential, where L* represents the lightness of the colour from 0 to 100 (black to white), a* represents how much red versus green (positive value at x axis means the colour inclines to be red and negative value at x axis means the colour inclines to be green), and b* represents how much yellow versus blue (positive value at y axis means the colour inclines to be yellow and negative value at y axis means the colour inclines to be blue).36Fig. 8 demonstrates that the lightness (L*) varies with the increase of voltage applied to the film. Three films with different absorption values are measured for both polymers, and the a* values and b* values in neutral states and oxidized states are also marked in the graph. For PBDBT, when the voltage rises, lightness (L*) firstly stays steady, then climbs to a stable stage, meaning that the colour of film turns to be light and transparent. a* turns from a big positive value to a negative value, in other words, red turns to be green, while b* shows a contrary trend, increasing from a big negative value to a small negative value, that means the color changes from blue to yellow. As shown in Fig. 8(a2) and (b2), it can be observed the trend how a* and b* change with the voltage. When the voltage increases, a* and b* go from the fourth quadrant to third quadrant. As a result, the film changes from rosybrown to transparent grey.
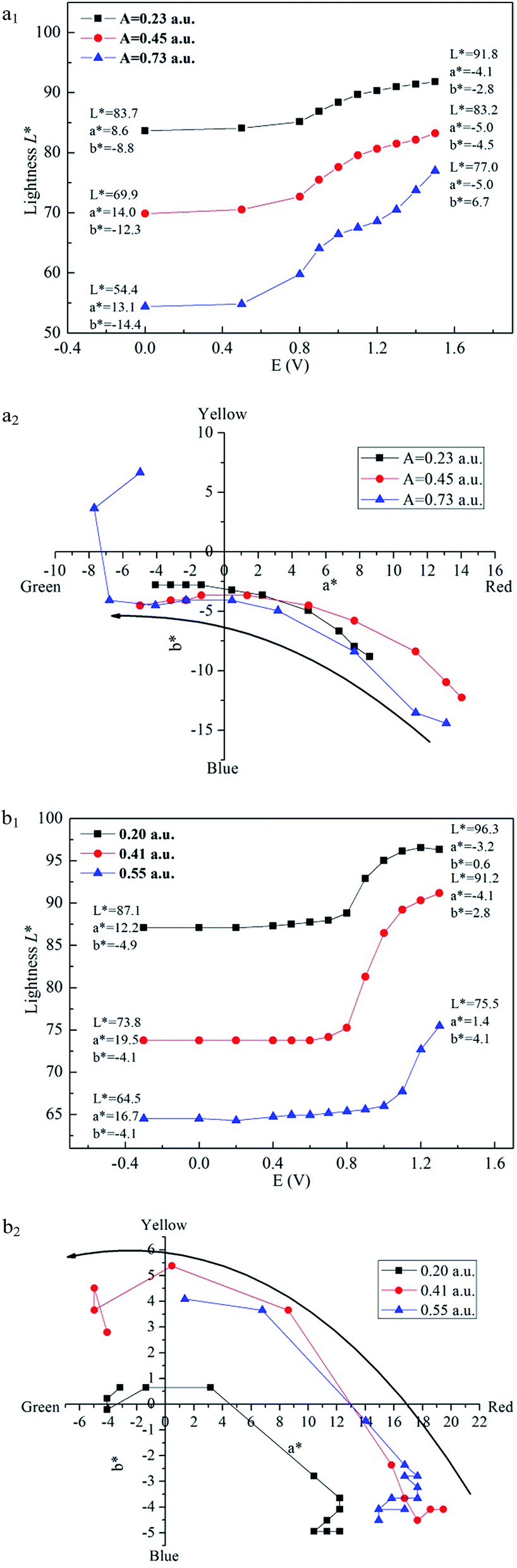 | ||
| Fig. 8 Plot of lightness as a function of applied voltage of PBDBT (a1) and PIDBT (b1) and the a*–b* graphs of PBDBT (a2) and PIDBT (b2). | ||
In addition, we can get the result that the thinner the film is, the lighter it is. When the film thickness of PBDBT changes from 0.23 a.u. (532 nm) to 0.45 a.u. and to 0.73 a.u., the lightness is 83.7, 69.9, 54.4 in neutral state and 91.8, 83.2, 77.0 in oxidized state, respectively (Fig. 8(a1)). There exist a significant increase, indicating that film thickness is an important parameter and should be considered carefully when people want to tune the film lightness.
Similar with PBDBT, the lightness of PIDBT film firstly keeps stable, and then has a dramatic increase when the voltage is nearly 1.0 V. a* value gets smaller, while b* value gets bigger (Fig. 8(b1)). When the film thickness is 0.20 a.u. (531 nm), the L*, a*, b* changes from 87.1, 12.2, −4.9 to 96.3, −3.2, 0.6, respectively. When the film turns thicker, the lightness turns darker (Fig. 8(b1)), with the same tendency as that of PBDBT.
Thermal gravimetric analysis of PBDBT and PIDBT
The molecular weight and the polydispersity of polymers are the important parameters for electrochromic applications. A high molecular weight favours the performance of electrochromic device in several aspects, including the improvement of the adhesion to the substrate, the resistant to the dissolution in the supporting electrolyte, and the maintenance of a high optical contrast after repeatedly electrochromic switching.33 There are three primary parameters characterizing the molecular weight of the polymer, the weight-average molecular weight (Mw), the number-average molecular weight (Mn), and the polydispersity (Mw/Mn) of the polymers. The molecular weights of the polymers against polystyrene standards were determined by gel permeation chromatography (GPC) taking tetrahydrofuran as the eluent phase. The GPC results showed that the Mw, Mn and the Mw/Mn data were 16.5 kDa, 7.6 kDa and 2.17 for PBDBT, respectively, and were 14.8 kDa, 7.8 kDa and 1.90 for PIDBT, respectively.To evaluate the thermal stability of the two copolymers, thermal gravimetric (TG) analysis experiment was carried out. The slight mass loss found in both polymers between 50 °C and 100 °C might be caused by the evaporation of the trace amounts of water or organic solvents contained in the polymers (Fig. 9). As shown in Fig. 9(a), PBDBT begins to decompose when the temperature reaches to 320 °C, and PIDBT begins to decompose when the temperature is nearly 460 °C. They both have a high initial decomposition temperature, and PIDBT is more stable than PBDBT with temperature rising. In order to more conveniently compare the thermal stabilities of two polymers, differential thermal gravity (DTG) curves were also shown in Fig. 9, from which the temperature points with the maximum degradation rates are found to be 356.6 °C and 489.6 °C, for PBDBT and PTDBT, respectively. The results also showed that PTDBT has better stability than that of PBDBT, which might due to the multi benzene structure of PTDBT backbone. The data revealed that both polymers had enough thermal stability for electrochromic applications.35
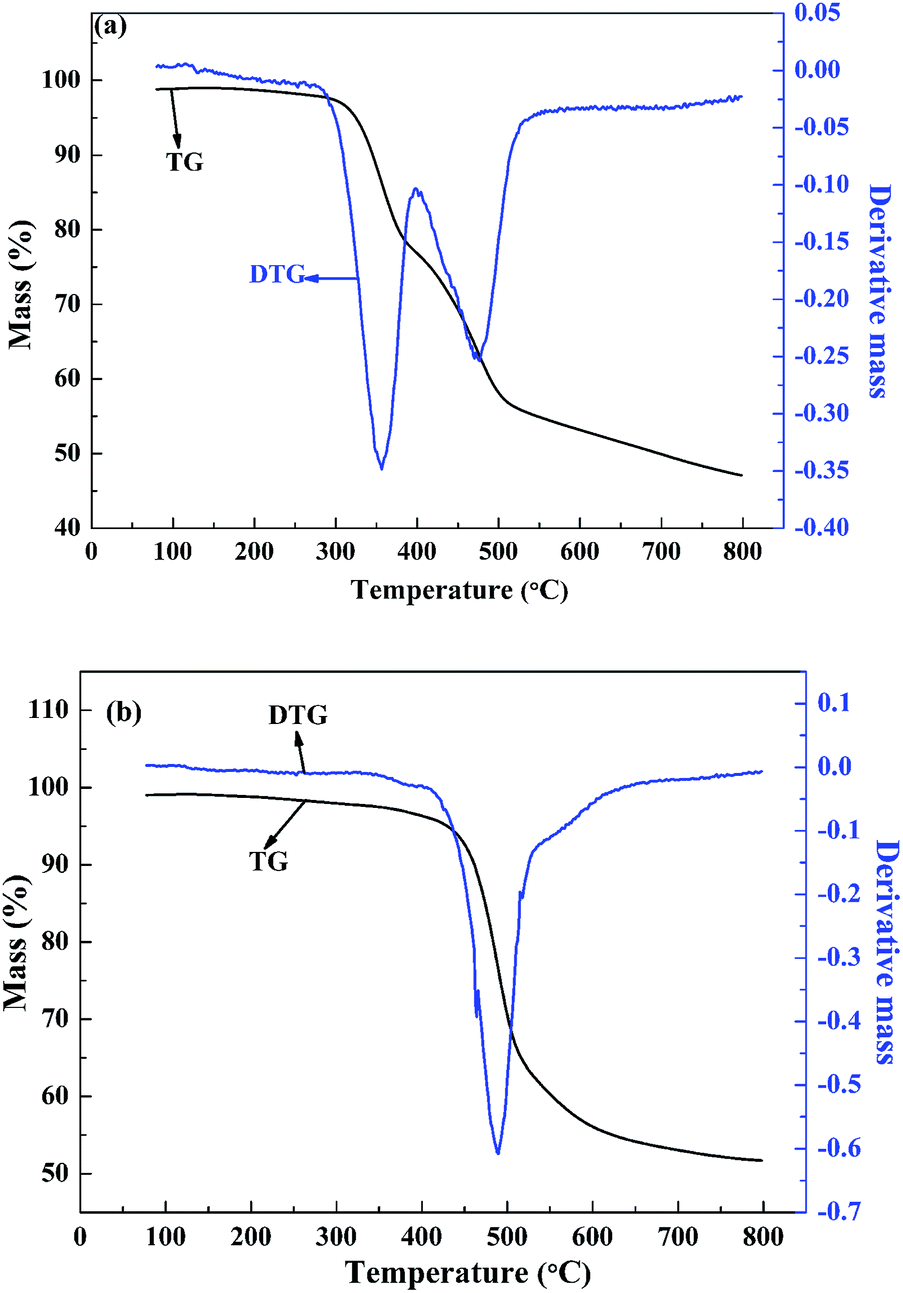 | ||
| Fig. 9 TG and DTG curves of PBDBT (a) and PIDBT (b). | ||
Conclusion
In summary, we have successfully synthesized two new copolymers, PBDBT and PIDBT through Stille coupling reaction. When oxidized, PBDBT turns from rosybrown to transparent grey, while for PIDBT, it turns from pale violet red in neutral state to transparent light grey in oxidized state. The band gap of both copolymers are 1.90 eV and 1.93 eV for PBDBT and PIDBT, respectively. They show fast response time in UV-Vis-NIR region, and also have satisfactory optical contrast. The optical contrasts in long wavelength region are very high. In addition, they have a high coloration efficiency at longer wavelength, the coloration efficiency of PBDBT at 1500 nm is 144.70 cm2 C−1, and the coloration efficiency of PIDBT at 1560 nm is 222.00 cm2 C−1. The challenges we will overcome are how to enhance the solubility of the copolymers and trying to reduce the band gaps. In summary, they both can be good candidates for electrochromic applications.Acknowledgements
The work was financially supported by the National Natural Science Foundation of China (51473074, 31400044), the General and Special Program of the postdoctoral science foundation China (2013M530397, 2014T70861), and the Natural Science Foundation of Shandong Province (ZR 2014JL009).Notes and references
- A. P. Kulkarni, C. J. Tonzola, A. Babel and S. A. Jenekhe, Chem. Mater., 2004, 16, 4556–4573 CrossRef CAS.
- A. J. Heeger, Solid State Commun., 1998, 107, 673–679 CrossRef CAS.
- J. Wang, G. Yu, G. Srdanov and A. Heeger, Org. Electron., 2000, 1, 33–40 CrossRef CAS.
- R. J. Mortimer, A. L. Dyer and J. R. Reynolds, Displays, 2006, 27, 2–18 CrossRef CAS.
- S. Celebi, A. Balan, B. Epik, D. Baran and L. Toppare, Org. Electron., 2009, 10, 631–636 CrossRef CAS.
- X. Fu, C. Jia, S. Wu, X. Weng, J. Xie and L. Deng, Synth. Met., 2014, 188, 104–110 CrossRef CAS.
- G. Gunbas and L. Toppare, Chem. Commun., 2012, 48, 1083–1101 RSC.
- Y. Li and J. B. Lagowski, Opt. Mater., 2010, 32, 1177–1187 CrossRef CAS.
- F. Larmat, J. Soloducho, A. R. Katritzky and J. R. Reynolds, Synth. Met., 2001, 124, 329–336 CrossRef CAS.
- G. Yu and A. J. Heeger, Synth. Met., 1997, 85, 1183–1186 CrossRef CAS.
- X. Liu, Y. Sun, B. B. Y. Hsu, A. Lorbach, L. Qi, A. J. Heeger and G. C. Bazan, J. Am. Chem. Soc., 2014, 136, 5697–5708 CrossRef CAS PubMed.
- C. DuBois, K. A. Abboud and J. R. Reynolds, J. Phys. Chem. B, 2004, 108, 8550–8557 CrossRef CAS.
- C. L. Chochos and S. A. Choulis, Prog. Polym. Sci., 2011, 36, 1326–1414 CrossRef CAS.
- J. Yu, H. Tan, F. Meng, K. Lv, W. Zhu and S. Su, Dyes Pigm., 2016, 131, 231–238 CrossRef CAS.
- A. Balan, G. Gunbas, A. Durmus and L. Toppare, Chem. Mater., 2008, 20, 7510–7513 CrossRef CAS.
- A. Balan, D. Baran, G. Gunbas, A. Durmus, F. Ozyurt and L. Toppare, Chem. Commun., 2009, 6768–6770 RSC.
- S. C. Price, A. C. Stuart, L. Yang, H. Zhou and W. You, J. Am. Chem. Soc., 2011, 133, 4625–4631 CrossRef CAS PubMed.
- H. Pan, Y. Li, Y. Wu, P. Liu, B. S. Ong, S. Zhu and G. Xu, J. Am. Chem. Soc., 2007, 129, 4112–4113 CrossRef CAS PubMed.
- J. Hou, M. H. Park, S. Zhang, Y. Yao, L. M. Chen, J. H. Li and Y. Yang, Macromolecules, 2008, 41, 6012–6018 CrossRef CAS.
- X. Zhang, L. Chen, G. Wang, Z. G. Zhang, Y. Li and P. Shen, Synth. Met., 2016, 211, 121–131 CrossRef CAS.
- N. Yin, L. Wang, Y. Ma, Y. Lin, J. Wu, Q. Luo, H. B. Yang, C. Q. Ma and X. Zhao, Dyes Pigm., 2015, 120, 299–306 CrossRef CAS.
- W. T. Neo, Q. Ye, T. T. Lin, S. J. Chua and J. Xu, Sol. Energy Mater. Sol. Cells, 2015, 136, 92–99 CrossRef CAS.
- M. Wang, X. Hu, L. Liu, C. Duan, P. Liu, L. Ying, F. Huang and Y. Cao, Macromolecules, 2013, 46, 3950–3958 CrossRef CAS.
- W. Tang, D. Huang, C. He, Y. Yi, J. Zhang, C. Di, Z. Zhang and Y. Li, Org. Electron., 2014, 15, 1155–1165 CrossRef CAS.
- G. Hızalan, A. Balan, D. Baran and L. Toppare, J. Mater. Chem., 2011, 21, 1804–1809 RSC.
- P. H. Aubert, M. Knipper, L. Groenendaal, L. Lutsen, J. Manca and D. Vanderzande, Macromolecules, 2004, 37, 4087–4098 CrossRef CAS.
- Y. Liu, M. Wang, J. Zhao, C. Cui and J. Liu, RSC Adv., 2014, 4, 52712–52726 RSC.
- W. T. Neo, L. M. Loo, J. Song, X. Wang, C. M. Cho, H. S. O. Chan, Y. Zong and J. Xu, Polym. Chem., 2013, 4, 4663–4675 RSC.
- S. Debnath, A. Bedi and S. S. Zade, Poly. Chem., 2015, 6, 7658–7665 RSC.
- C. L. Gaupp, D. M. Welsh and J. R. Reynolds, Macromol. Rapid Commun., 2002, 23, 885–889 CrossRef CAS.
- B. Wang, J. S. Zhao, C. Cui, R. Liu, J. Liu, H. Wang and H. Liu, Electrochim. Acta, 2011, 56, 4819–4827 CrossRef CAS.
- H. Zhao, D. Tang, J. Zhao, M. Wang and J. Dou, RSC Adv., 2014, 4, 61537–61547 RSC.
- T. Lee, C. A. Landis, B. M. Dhar, B. J. Jung, J. Sun, A. Sarjeant, H. J. Lee and H. E. Katz, J. Am. Chem. Soc., 2009, 131, 1692–1705 CrossRef CAS PubMed.
- W. Yang, J. Zhao, C. Cui, Y. Kong, X. Zhang and P. Li, J. Solid State Electrochem., 2012, 16, 3805–3815 CrossRef CAS.
- Z. Xu, M. Wang, J. Zhao, C. Cui, W. Fan and J. Liu, Electrochim. Acta, 2014, 125, 241–249 CrossRef CAS.
- C. M. Amb, A. L. Dyer and J. R. Reynolds, Chem. Mater., 2011, 23, 397–415 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |