
PTX encapsulated by an XG–DOX conjugate for combination therapy against multi-drug resistance
Zhuli Huang†
a,
Xuan Xie†a,
Jean Felix Mukerabigwia,
Chang Wanga,
Shufang Wangb,
Wang Xiao*a,
Xueying Huanga and
Yu Cao*a
aKey Laboratory of Pesticide and Chemical Biology (Ministry of Education), College of Chemistry, Central China Normal University, Wuhan 430079, P. R. China. E-mail: caoyu@mail.ccnu.edu.cn; xiaowang@mail.ccnu.edu.cn
bBlood Transfusion Department, The General Hospital of the People’s Liberation Army, Beijing 100853, China
First published on 21st October 2016
Abstract
So far, several anti-cancer drugs have shown low efficacy in the clinic because of multi-drug resistance (MDR), which always leads to the failure of chemotherapy. In addition, high doses of these chemotherapeutic drugs such as doxorubicin (DOX) and paclitaxel (PTX) result in high toxicity. Considering the above problems, a kind of dual drug loading onto a conjugate was designed, which makes good use of two drugs. In this new nano-drug delivery system, DOX works as a hydrophobic core and xyloglucan (XG) works as a hydrophilic shell to form stable nanoparticles in aqueous solution. Thereafter, PTX is encapsulated by this XG–DOX conjugate (PTX nano-DDS). Results have demonstrated that PTX nano-DDS possesses outstanding advantages over ordinary systems, including precise control over the molar ratio of the drugs and high hepatic targeting. Moreover, through the combination of two drugs, the system can maximize efficacy through synergism between PTX and DOX. It also has minimal side effects and is effective against MDR cancer cells. This kind of system is therefore practical for development as a new type of targeted dual drug delivery system in combination therapy.
1. Introduction
According to rough statistics, about 5 million people die from cancer annually all over the world.1 Although it can be treated using chemotherapy, not every person can be cured because of chemotherapy’s high risks and side effects. The possibility of cancer recrudescence also cannot be ignored. What is worse is multi-drug resistance,2,3 which leads directly to the failure of chemotherapy. Considering the above problems, this study proposed a concept called ‘combination therapy’, a new approach to fight cancer cells with two or more drugs through their synergism instead of one single drug.4,5 One of its prominent advantages is that it can inhibit cancer-drug resistance by damaging or killing the cancerous cells at their different stages of growth. Doxorubicin (DOX) and paclitaxel (PTX) were chosen as model drugs. The most common chemotherapeutics in the clinic, DOX is a kind of anthracycline medicine which has a mechanism of action against DNA, and PTX is hydrophobic which is beneficial for the formation of stable microtubules which can disrupt a cell’s mitosis and proliferation and cause the apoptosis of cancer cells. Combination therapy of DOX and PTX has been proven in the clinic to have better efficacy in terms of cancer recurrence rates. Besides, carriers of drugs and a release kinetics study of multi-drug payloads are worthy of attention, as well as the suitable formulation of dual drugs.In recent years, polymeric drug conjugates have become more and more popular as the choice of drug carrier. Their superior properties such as low toxicity, high permeability, good retention and bio-availability have been demonstrated in several studies. Clinical trials have proven their ability to promote synergism among drugs and inhibit cancer recrudescence efficiently. Most important is that polymeric drug conjugates can control the molar ratio of the drugs precisely.6,7 However, the variability of interactions between the drugs or the drug and the polymer as well as steric hindrance between the drug molecules tend to affect the precise radiometric delivery and synchronized multi-drug release of a single carrier.8,9 A new method is proposed in this study which loads different molar ratios of dual drugs onto a single carrier.
The tripeptide Gly-Leu-Gly plays an important role in this study. It is used as a spacer in case of binding between drugs. It is stable in the bloodstream and degrades in cancer cells which contain a variety of lysosomal enzymes. Xyloglucan (XG) functions as the drug carrier in this experiment. It is a polysaccharide which consists of a (1 → 4)-β-D-glucan main chain with (1 → 6)-α-D-xylose branches which are partially substituted by (1 → 2)-β-D-galactoxylose.10 Since it is natural, water-soluble, biodegradable and nontoxic, XG is widely used in the medicinal industry, especially as the hydroxyl groups of it are used as anchoring points for anti-tumor drugs.11,12 The XG–DOX polymer, with a hydrophobic core (DOX) and a hydrophilic shell (XG), self-assembles and enables the formation of stable nanoparticles in aqueous solution. It was expected that the hydrophobic anti-cancer drug PTX could be encapsulated into the polymeric micelle system efficiently, and this delivery system could possess excellent MDR reversing capabilities. The therapeutic effect on drug resistant variants of hepatoma cells (HepG2/DR) was evaluated in vitro and in vivo. This approach maximizes the combination of drugs’ efficacy as a result of the precise control of the dosage.
2. Materials and methods
2.1. Materials
Xyloglucan (XG) was extracted from tamarind powder (food grade) purchased from Henan Anrui Biotechnology Co., Ltd (China). Doxorubicin (DOX) was purchased from Chuangcheng Pharmaceutical (Wuhan) Ltd (China). Boc-Gly-Leu-Gly-Osu was purchased from GL Biochem (Shanghai) Ltd (China). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and a lysosome separation ELISA kit was purchased from the Haling Bio-technology Co., Shanghai. Paclitaxel (PTX) was purchased from Xi’an Sanjiang Bio-Engineering Co., Ltd (China). Free PTX solution was prepared according to the commercial formulation of Taxol. All the other chemicals were analytical grade and used without any further purification unless otherwise stated.2.2. Preparation and purification of XG
XG was extracted from tamarind powder and its solution (2 wt%) was prepared by dissolving the tamarind powder in distilled water. The solution was stirred in a heating-water bath for two hours and kept overnight. Then the water-insoluble protein fraction was removed by centrifugation at 5000 rpm for 30 min and absolute ethanol (twice the volume of the solution) was added before suction filtration with a sand core funnel (G4, pore size, 5–15 μm). Afterwards, the filtrate solution was lyophilized to gain pure XG for further use. The molecular weight of XG was about 700 kDa measured by GPC (Prominence GPC, Shimadzu Co., Japan).2.3. Preparation of peptide–DOX derivatives
DOX (1 g, 1.84 mmol) and Boc-Gly-Leu-Gly-Osu (0.75 g, 1.70 mmol) were dissolved in DMF, and the solution was stirred while DEPC (0.4 g, 0.25 mmol) and triethylamine (TEA) (0 °C, 0.3 ml) were added. The whole solution was stirred for 0.5 hours and kept at room temperature overnight to react. Then the solvent was evaporated under vacuum conditions and ethyl acetate was added to dissolve the residue. 10% citric acid solution (3 × 5 ml) and saturated sodium bicarbonate (3 × 5 ml) were added and the solution extracted with ethyl acetate (2 × 5 ml). Then the ethyl acetate extracts were evaporated under vacuum conditions, and the residue was purified by column chromatography on silica. The collected fraction was dried using MgSO4. The Boc-Gly-Leu-Gly–DOX derivative was obtained after removal of the solvent. Boc-Gly-Leu-Gly–DOX (0.1 g) was dissolved in DMF (2 ml), and trifluoroacetic acid (TFA) (0.2 ml) was added. The reaction mixture was stirred at room temperature for one hour. The solvent was evaporated under vacuum conditions and the residue was dissolved in methanol (5 ml). The solution was filtered and the filtrate was evaporated and eventually the Boc-Gly-Leu-Gly–DOX conjugated polymer was obtained.2.4. Preparation of the XG–DOX polymer
XG (2 g, 12.3 mmol) and DMAP (0.15 g, 1.2 mmol) were dissolved in DMSO/pyridine (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) solution (20 ml). 4-Nitrophenyl chloroformate (0.9 g, 4.4 mmol) was added. The mixture was stirred at 0 °C for 4 hours and precipitated using absolute ethanol. A white solid was obtained and washed three times with absolute ethanol. Then the obtained xyloglucan–COO(C6H4)NO2 was dried under vacuum conditions. The carbonate content was determined by UV analysis after the hydrolysis of activated XG in NaOH.
:1, v/v) solution (20 ml). 4-Nitrophenyl chloroformate (0.9 g, 4.4 mmol) was added. The mixture was stirred at 0 °C for 4 hours and precipitated using absolute ethanol. A white solid was obtained and washed three times with absolute ethanol. Then the obtained xyloglucan–COO(C6H4)NO2 was dried under vacuum conditions. The carbonate content was determined by UV analysis after the hydrolysis of activated XG in NaOH.
Xyloglucan–COO(C6H4)NO2 (2 g, 1.3 mmol), Gly-Leu-Gly–DOX (2 g) and TEA (0.1 ml) were dissolved in anhydrous DMSO in turn. The mixture was separated by precipitation in absolute ethanol after a 48 hour reaction in the dark. The obtained solid was washed, dried and purified by preparative GPC (Sephadex G25) with water as the eluent and freeze-drying. The DOX content was determined by UV analysis in water.
2.5. Preparation and characterization of PTX nano-DDS
XG–DOX (100 mg) was dissolved in DMSO (10 ml) and dialyzed (MWCO 8000 Da) against excessive deionized water at 4 °C for 3 days. The water was exchanged every 8 hours. The collected substance was filtrated using a 0.45μm-membrane and the nanoparticles were obtained after freeze-drying. Preparation of PTX nano-DDS was conducted through the loading of PTX into the XG–DOX polymer. PTX (10 mg) was dissolved in anhydrous DMSO (10 ml) so that the molar ratio of triethylamine and PTX was 2:1. XG–DOX polymer (50 mg) was added and the mixture was stirred overnight under cold conditions. Then the solution was dialyzed against excessive deionized water at 4 °C for 3 days and the water was exchanged every 8 hours. The nanoparticles self-assembled through the direct dispersal of the organic layer into the water layer. After that, extensive dialysis against deionized water was carried out to remove the unencapsulated DOX and triethylamine. Eventually, PTX nano-DDS was obtained after filtration using a 0.45μm-membrane, centrifugation, separation and lyophilization, which lasted for 3 days.
The PTX entrapment proportion was calculated as follows:

2.6. In vitro studies
:20:30 v/v/v, pH 5.0, 0.2 M) was exchanged at regular intervals, an Extend-C18 column (4.6 × 250 mm i.d., 5 μm) was used and the flow rate was 0.5 ml min−1. The content of DOX in the solution was determined by UV spectroscopy at 245 nm, and the content of PTX was determined by UV spectroscopy at 360 nm.15,162.7. In vivo studies
All the studies implemented on animals were in accordance with the “Guidelines for the Care and Use of Laboratory Animals” published by the National Institutes of Health (NIH Publication no. 85-23, revised 1985). This study was approved by the Ethics Committee of Central China Normal University.Five groups (36 per group) of mice were injected with free DOX, free PTX, XG–DOX, XG–DOX with free PTX (1:1) and PTX nano-DDS (14 mg kg−1), respectively. 24 hours later, a 0.5 ml blood sample was obtained from the retro-orbital venous plexus puncture of the tumor-bearing mice. The livers, hearts, kidneys and tumors were separated immediately and washed with Na2HPO4 buffer (10 mM), followed by homogenization with ethyl acetate solvent. The drug was extracted after being incubated with acidic isopropanol at 4 °C for 4 hours. Then the mixture was agitated in a vortex mixer for 1 min and subsequently centrifuged. PTX and DOX concentrations were determined by HPLC. Free or released drug was extracted and determined without incubation by HPLC as mentioned above.17,18
2.8. Data analysis
Data was expressed as means ± standard deviations of multireplicated determinations. The Student t test was used to compare the differences between the means of two groups at the same time. A value was considered to be significant if p < 0.05.3. Results
3.1. Preparation and characterization of XG–DOX
Before attaching doxorubicin (DOX) to xyloglucan (XG), XG was first activated using a polysaccharide mixed with 4-nitrophenyl chloroformate. Then, Boc-Gly-Leu-Gly-Osu was used as a spacer to link DOX and XG. The relevant products have been characterized by FTIR and 1H-NMR spectroscopy and the results are shown in Fig. 1 and 2, respectively. The absorption peaks at 2860 cm−1 and 2929 cm−1 in the FTIR spectrum of XG–DOX indicate the conjugation between XG and DOX. The peak at 1452 cm−1 might be assigned to the absorption of PTX in nano-DDS. In Fig. 2, compared to the spectrum of XG, the new peaks at 8.31 ppm and 7.51 ppm for XG–COO(C6H4)NO2 can be identified as the hydrogens of benzene. This demonstrates the activation of XG. In addition, the peak at 1.10 ppm for XG–DOX was considered as the methyl group on DOX, which confirms the conjugation between XG and DOX. | ||
| Fig. 1 FTIR spectra of xyloglucan (XG), doxorubicin (DOX), xyloglucan–doxorubicin (XG–DOX) and PTX nano-DDS. | ||
 | ||
| Fig. 2 1H NMR spectra of xyloglucan (XG), xyloglucan–COO(C6H4)NO2 (XG–NPC) and xyloglucan–doxorubicin (XG–DOX). | ||
3.2. Characterization of PTX nano-DDS
PTX nano-DDS was characterized by its particle size and drug-encapsulation efficiency. The measurement results of the DLS analysis (Fig. 3) show that PTX nano-DDS has a larger size after encapsulating PTX. The diameter reaches up to 100.1 nm with a PDI of 0.262. The zeta-potential of PTX nano-DDS was 27.8 ± 2.5 mV. These nanoparticles are able to enter into tumor tissues more easily due to the enhanced permeability and retention (EPR) effect. Moreover, the content of PTX reaches up to 18.5% and the entrapment efficacy reaches up to 82%. | ||
| Fig. 3 The DOX nano-DDS and PTX nano-DDS size distributions determined by DLS analysis (left) and the TEM micro-graph of PTX nano-DDS (right). | ||
3.3. Drug released from PTX nano-DDS
The release proportion of free DOX, free PTX and nano-DDS in collagenase IV and serum was evaluated as is shown in Fig. 4. The graphs demonstrate that for the same kind of drug, the percentage released in serum is much smaller than in collagenase IV, so that the release is negligible. The slight release of the drugs indicates that they are stable in plasma circulation. In collagenase IV, the proportion of drug-release increases over time and all three kinds of drug system are released by more than 50% after 15 hours. Notably, nano-DDS reaches up to 80% after 48 h, beyond the release proportions of free DOX and free PTX.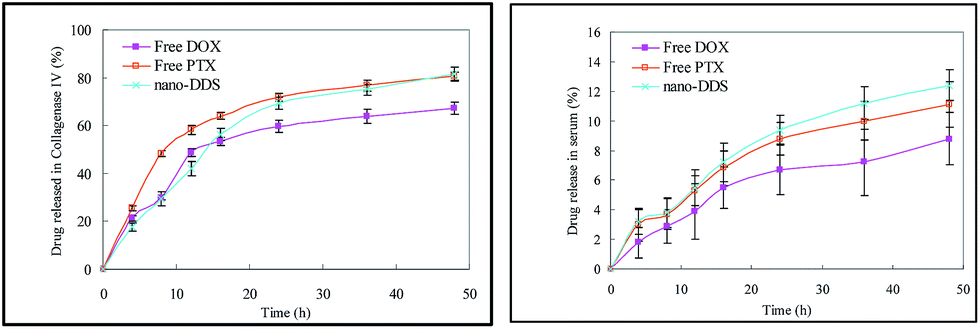 | ||
| Fig. 4 Release curves of free DOX, free PTX and PTX nano-DDS in two kinds of medium (collagenase IV and serum). Data are given as mean ± SD (**p < 0.05). | ||
3.4. In vitro cytotoxicity of PTX nano-DDS against drug-resistant HepG2 cells
The cytotoxicity of PTX nano-DDS against HepG2/DOX cells was determined by MTT. The results are shown in Table 1. Compared to parental HepG2 cells, drug-resistant cells apparently have a higher IC50 value. DOX and PTX have the highest IC50 values, so it’s not appropriate to use free drugs when it comes to drug-resistant cells. It is found that the combination of XG–DOX and PTX apparently lowers the IC50, which means that the combination therapy possesses an increased efficacy, and when PTX is encapsulated in XG–DOX instead being free PTX, the IC50 is again lower. Additionally, compared to if the formulation has equal amounts of XG–DOX and PTX, when the amount of PTX or XG–DOX is increased, the IC50 changes. Moreover, a change of PTX is more effective than that of XG–DOX because the IC50 value of XG–DOX/PTX (1:1) is 0.374 ± 0.018/0.374 ± 0.015, and that of XG–DOX/PTX (1:4) is 0.499 ± 0.045/1.998 ± 0.017, while that of XG–DOX/PTX (4:1) is 1.052 ± 0.029/0.261 ± 0.024. Generally speaking, the XG–DOX/PTX (1:1) system has the best efficacy and synergism.
| Formulation | IC50 of HepG2 (μmol l−1) | IC50 of HepG2/DOX (μmol l−1) |
|---|---|---|
| DOX | 0.159 ± 0.009 | 15.843 ± 0.126 |
| PTX | 0.189 ± 0.013 | 6.383 ± 0.111 |
| XG–DOX | 0.062 ± 0.008 | 1.052 ± 0.029 |
| XG–DOX + PTX (1:1) |
0.059 ± 0.011/0.059 ± 0.011 | 1.002 ± 0.039/1.002 ± 0.039 |
| XG–DOX/PTX (1:4) |
0.047 ± 0.012/0.188 ± 0.015 | 0.499 ± 0.045/1.998 ± 0.017 |
| XG–DOX/PTX (1:2) |
0.038 ± 0.006/0.076 ± 0.009 | 0.464 ± 0.021/0.928 ± 0.032 |
| XG–DOX/PTX (1:1) |
0.028 ± 0.007/0.028 ± 0.005 | 0.374 ± 0.018/0.374 ± 0.015 |
| XG–DOX/PTX (2:1) |
0.052 ± 0.009/0.026 ± 0.004 | 0.926 ± 0.026/0.463 ± 0.016 |
| XG–DOX/PTX (4:1) |
0.064 ± 0.007/0.016 ± 0.006 | 1.052 ± 0.029/0.261 ± 0.024 |
3.5. In vivo pharmacokinetic study and bio-distribution in tumor-bearing mice
After injection of different drugs (14 mg kg−1), the distribution of the drugs in the mice’s bodies is measured. The results are shown in Fig. 5 and Table 2 as follows. In the blood, the concentrations of the drugs decrease over time, with the rate of decrease of the free drugs being much quicker than that of the drug in nano-DDS. This demonstrates that the free drugs degrade quicker in circulation than drugs in nano-DDS. The same conclusion can be observed from Table 2, in which nano-DDS has a larger area under the curve (AUC) than the free drugs. | ||
| Fig. 5 Bio-distribution of drug concentration in tumor-bearing mice after a single dose of 14 mg kg−1 of free DOX, free PTX, XG–DOX, XG–DOX with free PTX (1:1) and nano-DDS. Data is given as mean ± SD (**p < 0.05). | ||
| Tissue | Free DOX | XG–DOX | Free PTX | Nano-DDS | XG–DOX + PTX | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AUC, μg g−1 h | MRT, h | t1/2, h | AUC, μg g−1 h | MRT, h | t1/2, h | AUC, μg g−1 h | MRT, h | t1/2, h | AUC, μg g−1 h | MRT, h | t1/2, h | AUC, μg g−1 h | MRT, h | t1/2, h | |
| Blood | 3.591 | 4.023 | 2.789 | 21.534 | 2.376 | 1.584 | 2.9760 | 0.034 | 0.024 | 27.676 | 2.377 | 1.647 | 12.255 | 1.205 | 0.804 |
| Heart | 74.07 | 10.21 | 7.075 | 6.6980 | 1.628 | 1.085 | 15.763 | 6.564 | 4.550 | 8.6050 | 1.628 | 1.129 | 11.232 | 4.096 | 2.731 |
| Kidney | 3.613 | 0.035 | 0.024 | 43.494 | 18.51 | 12.34 | 22.987 | 1.733 | 1.201 | 55.890 | 18.52 | 12.84 | 33.241 | 10.13 | 6.753 |
| Liver | 91.53 | 9.825 | 6.810 | 432.30 | 23.78 | 15.85 | 84.049 | 5.573 | 3.863 | 555.47 | 23.78 | 16.48 | 258.18 | 14.68 | 9.787 |
| Tumor | 1.359 | 4.534 | 1.961 | 803.58 | 443.8 | 295.9 | 10.431 | 1.522 | 1.055 | 1032.6 | 443.7 | 307.6 | 407.01 | 222.7 | 148.5 |
The bio-distribution of the drugs in some organs of the tumor-bearing mice have also been measured, including the liver, kidney and heart. The general trend is that the concentrations of all the drugs decrease over time, and the content of drugs in these organs is very low after 24 hours. Therefore these drugs can be metabolized well in the body. The mean residence time (MRT) and t1/2 of the drugs are both less than 12 hours. This means that these drugs have low toxicity residues for the organs above. However, it also has been found that the concentration of drugs from nano-DDS in the liver increases at the beginning, and all the drugs’ concentrations apparently decrease more slowly in the liver than in other organs. This reflects that the targeting properties of nano-DDS are better because its concentration is the highest throughout the process.
With regards to the tumor, the situation is very different. Though free DOX and free PTX still degrade quickly over 10 hours, the concentration of drugs from nano-DDS increase at the beginning and reach a maximum (nano-DDS: 51.988 μg g−1) at the eighth hour, which indicates the best efficacy at this time. It also starts to decrease later but the concentration is still higher than the initial value. In addition, the values of AUC, MRT and t1/2 of nano-DDS are much higher than for the other formulations of the drugs in the tumor. So, nano-DDS can stay in a tumor for a much longer period of time than the other formulations which enables it to work to its fullest capacity. The advantages of using nano-DDS are shown significantly in the tumor results. Moreover, nano-DDS has a larger AUC (1032.6 μg g−1 h) than XG–DOX (803.58 μg g−1 h), which indicates that nano-DDS has a better efficacy.
3.6. In vivo anti-tumor efficacy
In order to observe the effect of the drugs against MDR cells, experiments were conducted on mice implanted with HepG2/DOX cells. The mice were divided into four groups and each was injected with one kind of drug per week, and the tumor volume size and alive mice numbers were recorded in the following weeks. The results are shown in terms of tumor volume changes (Fig. 6) and alive mice numbers (Fig. 7) as follows. In the first week, the sizes of the tumors were similar and in the second week, they started to differ from each other. The curve trends demonstrate that XG–DOX and nano-DDS have superior abilities to suppress the growth of tumors compared to saline, XG–DOX and free DOX. Especially nano-DDS, which had the best efficacy and stopped the tumor’s size from increasing by more than 520 mm3. Hence, the growth of tumors can be restrained well by using nano-DDS. From Fig. 7 a similar conclusion can be drawn. It was found that all the mice that were injected with saline or free DOX died within the first four weeks, while all the mice that were injected with XG–DOX, XG–DOX + PTX or nano-DDS were still alive. Also, the group injected with XG–DOX + PTX resulted in a dead mouse first, and a few days later the group injected with XG–DOX then exhibited its first dead mouse. | ||
| Fig. 6 Tumor volume changes of the xenografted nude mice bearing the HepG2/DOX tumors over three weeks. These mice were divided into five groups and each group was injected with one drug (4 mg kg−1) by tail vein injection for 4 doses (on days 0, 7, 14, and 21). | ||
 | ||
| Fig. 7 Alive mice number plots of the treated xenografted nude mice bearing the HepG2/DOX tumors. These mice were divided into five groups and each group was injected with one drug (4 mg kg−1) every week for a total of 4 doses (on days 0, 7, 14, and 21). | ||
However, there were still four mice injected with nano-DDS that were alive after all the mice injected with XG–DOX had died. On the fiftieth day, there was still one mouse which was alive which had been injected with nano-DDS. Apparently, nano-DDS helped to control the spread of the tumor excellently and improved the survival rate of the mice. It has significant advantages over other drugs, considering its ability to affect tumor resistance and prolong the life span.
4. Discussion
A creative drug-delivery system was designed in this study which encapsulated PTX in XG–DOX. Synergism between DOX and PTX maximized the efficacy of chemotherapy, compensating for the deficiencies of the use of one single drug. Considering that xyloglucan (XG) is one of the most promising carriers for macro-molecular drugs,20 it was used as the drug carrier in this system. There are many hydroxyl residues in XG that can encapsulate drugs within a hydrophilic shell and help the whole system to realize long-term retention in circulation. This can also explain the phenomenon that occurred in the experiments on cytotoxicity in vivo (Fig. 6 and 7). The two charts show that nano-DDS has a superior ability among all the drugs tested to suppress tumor growth and prolong the life span of mice. XG–DOX also has good efficacy compared to the free drug because it has XG as a hydrophilic shell and DOX as a hydrophobic core, and a structure similar to nano-DDS. This kind of system can achieve positive therapeutic efficacy through its special structure to enable the precise control of the ratio of drugs.21To find the ideal drug formulation, studies were conducted to measure the relevant IC50 values (Table 1). The results shown in the table demonstrate that the formulation when PTX was encapsulated in an equal amount of XG–DOX resulted in the best efficacy, which indicates that XG–DOX/PTX (1:1) has the most significant synergism. Moreover, compared to equal amounts of free PTX and XG–DOX, encapsulating PTX into XG–DOX obviously improved the drug-delivery rate because the drug will not degrade as quickly in circulation.22 According to the DLS results (Fig. 3), the size of the drug also has an effect. PTX nano-DDS is larger (100.1 nm) and it was found that a larger size is beneficial because smaller particles will react more easily with biological components and cause some side effects due to their larger surface.23,24 Particles whose sizes are less than 200 nm can accumulate in tumors through enhanced permeability and retention (EPR) effects.25,26 Moreover, the zeta-potential of PTX nano-DDS was 27.8 ± 2.5 mV. Nanoparticles with a zeta-potential of about 30 mV have been shown to be stable in suspension because the surface charge can prevent the aggregation of the particles.27,28
The tripeptide glycyl-L-leucyl-glycine (Gly-Leu-Gly) was used as the linker between DOX and XG. The XG–DOX polymer was synthesized and characterized using FTIR (Fig. 1) and 1H-NMR spectroscopy (Fig. 2). This peptide-linker can be hydrolyzed by lysosomal enzymes but can be stable in serum. A lot of enzymes exist in lysosomes including proteases which degrade the peptide-linker to achieve intracellular drug release.9,29 The release of the drugs in collagenase IV and serum were compared (Fig. 4). All the drugs were released so little in serum that it can be considered negligible compared to the release in collagenase IV. This indicates that the drugs are stable in serum rather than being hydrolyzed. In these two kinds of medium, free PTX is released the most quickly and more than PTX nano-DDS in collagenase IV. However, the ultimate released amount of PTX nano-DDS is still a little more than that of free PTX, which means that PTX nano-DDS can make more of the drugs accumulate in tumor cells under the same conditions. Once the drug is injected into the body it will be distributed with the circulation of the blood, through which the drug will enter into some organs.29,30 Considering this, it is necessary to conduct measurements on some important organs including the liver, kidney and heart to examine the toxic residues of the drugs. The results are presented in the form of a curve (Fig. 5). The general trend of the drugs’ concentration changes in the blood, liver, kidney and heart is a decrease over time. After 24 hours, the content of the drugs is so little that it can be ignored, except in the tumor. The situation in the tumor is completely different from the four systems before it. Though the concentrations of free DOX and free PTX still decrease very quickly at the beginning and almost drop to zero after four hours, the concentration of nano-DDS starts to increase at the beginning and keeps increasing over the following eight hours. Their ultimate concentrations are still higher than the initial values (14 mg kg−1). In addition, PTX nano-DDS has shown higher AUC, MRT and t1/2 values in the tumor (Table 2). This demonstrates that nano-DDS appears to have a significant targeting ability and little side effects on other organs. In all, this fresh approach to carrying dual drugs using the same conjugated polymer can be considered as a new opportunity in MDR cancer chemotherapy.
Acknowledgements
This research was supported by grants from the self-determined research of Central China Normal University (Fundamental Research Funds for the Central Universities CCNU15A02062), the China Spark Program (Ministry of Science and Technology, 2013GA740073), and the National Natural Science Foundation of China (51603081).References
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688–701 CrossRef CAS PubMed.
- I. Brigger, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2002, 54, 631–651 CrossRef CAS PubMed.
- Q. Wu, Z. Yang, Y. Nie, Y. Shi and D. Fan, Cancer Lett., 2014, 347, 159–166 CrossRef CAS PubMed.
- Z. Liu, Y. Jiao, Y. Wang, C. Zhou and Z. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1650–1662 CrossRef CAS PubMed.
- T. Sonia and C. P. Sharma, in Chitosan for biomaterials I, Springer, 2011, pp. 23–53 Search PubMed.
- Y. Bae, T. A. Diezi, A. Zhao and G. S. Kwon, J. Controlled Release, 2007, 122, 324–330 CrossRef CAS PubMed.
- C. Deng, Y. Jiang, R. Cheng, F. Meng and Z. Zhong, Nano Today, 2012, 7, 467–480 CrossRef CAS.
- S. Aryal, C. M. J. Hu and L. Zhang, Small, 2010, 6, 1442–1448 CrossRef CAS PubMed.
- S. Aryal, C.-M. J. Hu and L. Zhang, Mol. Pharmaceutics, 2011, 8, 1401–1407 CrossRef CAS PubMed.
- H. S. Mahajan, V. K. Tyagi, R. R. Patil and S. B. Dusunge, Carbohydr. Polym., 2013, 91, 618–625 CrossRef CAS PubMed.
- A. Mishra and A. V. Malhotra, J. Mater. Chem., 2009, 19, 8528–8536 RSC.
- T. Coviello, P. Matricardi and F. Alhaique, Expert Opin. Drug Delivery, 2006, 3, 395–404 CrossRef CAS PubMed.
- P. Sawant, I. Desai and A. Tappel, Biochim. Biophys. Acta, Spec. Sect. Enzymol. Subj., 1964, 85, 93–102 CAS.
- P. K. Smith, R. I. Krohn, G. Hermanson, A. Mallia, F. Gartner, M. Provenzano, E. Fujimoto, N. Goeke, B. Olson and D. Klenk, Anal. Biochem., 1985, 150, 76–85 CrossRef CAS PubMed.
- Y. Zhang, H. Wang, J. F. Mukerabigwi, M. Liu, S. Luo, S. Lei, Y. Cao, X. Huang and H. He, RSC Adv., 2015, 5, 71164–71173 RSC.
- H. S. Yoo and T. G. Park, J. Controlled Release, 2004, 100, 247–256 CrossRef CAS PubMed.
- A. De Marre, H. Soyez, E. Schacht, M. A. Shoaibi, L. W. Seymour and B. Rihova, J. Controlled Release, 1995, 36, 87–97 CrossRef.
- L. W. Seymour, K. Ulbrich, J. Strohalm, J. Kopeček and R. Duncan, Biochem. Pharmacol., 1990, 39, 1125–1131 CrossRef CAS PubMed.
- X. Duan, J. Xiao, Q. Yin, Z. Zhang, H. Yu, S. Mao and Y. Li, ACS Nano, 2013, 7, 5858–5869 CrossRef CAS PubMed.
- M. Monsigny, C. Petit and A.-C. Roche, Anal. Biochem., 1988, 175, 525–530 CrossRef CAS PubMed.
- Y.-I. Jeong, J.-B. Cheon, S.-H. Kim, J.-W. Nah, Y.-M. Lee, Y.-K. Sung, T. Akaike and C.-S. Cho, J. Controlled Release, 1998, 51, 169–178 CrossRef CAS PubMed.
- A. Gabizon, R. Catane, B. Uziely, B. Kaufman, T. Safra, R. Cohen, F. Martin, A. Huang and Y. Barenholz, Cancer Res., 1994, 54, 987–992 CAS.
- L. Mazzarino, G. Loch-Neckel, L. dos Santos Bubniak, F. Ourique, I. Otsuka, S. Halila, R. C. Pedrosa, M. C. Santos-Silva, E. Lemos-Senna and E. C. Muniz, Toxicol. Sci., 2015, 147, 104–115 CrossRef CAS PubMed.
- K. L. Aillon, Y. Xie, N. El-Gendy, C. J. Berkland and M. L. Forrest, Adv. Drug Delivery Rev., 2009, 61, 457–466 CrossRef CAS PubMed.
- Y. Cao, Y. Gu, H. Ma, J. Bai, L. Liu, P. Zhao and H. He, Int. J. Biol. Macromol., 2010, 46, 245–249 CrossRef CAS PubMed.
- I. Bertholon, H. Hommel, D. Labarre and C. Vauthier, Langmuir, 2006, 22, 5485–5490 CrossRef CAS PubMed.
- R. Xu, Particuology, 2008, 6, 112–115 CrossRef CAS.
- R. Singh and J. W. Lillard, Exp. Mol. Pathol., 2009, 86, 215–223 CrossRef CAS PubMed.
- L. Chen, X. Sha, X. Jiang, Y. Chen, Q. Ren and X. Fang, Int. J. Nanomed., 2013, 8, 73 Search PubMed.
- D. Kim, E. S. Lee, K. Park, I. C. Kwon and Y. H. Bae, Pharm. Res., 2008, 25, 2074–2082 CrossRef CAS PubMed.
Footnote |
| † Equal contributors to the work. |
| This journal is © The Royal Society of Chemistry 2016 |