
Spark plasma sintering route to synthesize aluminium doped zinc oxide
Abstract
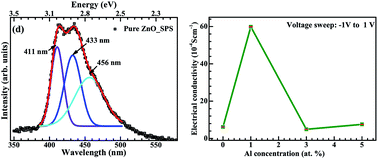
Aluminium doped zinc oxide is prepared using spark plasma sintering. The process leads to a tailored optical band gap extending into the visible region with increasing aluminium concentration. Microstructural, optical, and electrical characterization were carried out to investigate the effect of the sintering process and aluminium concentration on the grain size, band gap, and electrical conductivity. The delocalization of impurity energy states created during sintering, causes band gap narrowing and leads to an improved conductivity. The impurity states are primarily oxygen vacancies and zinc interstitials. Spark plasma sintering offers a promising approach to synthesize doped conductive metal oxides with control over the band gap and can be extended to a variety of other systems.
 Please wait while we load your content...
Please wait while we load your content...

