
Self-assembling ultrashort NSAID-peptide nanosponges: multifunctional antimicrobial and anti-inflammatory materials†

Abstract
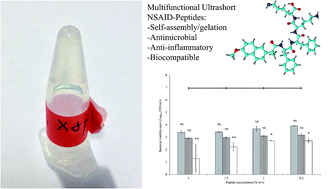
Peptide-based materials are receiving significant attention for use within biomedicine due to their high chemical and functional versatility enabling tailoring of their structure to replicate the properties of host tissue and the extracellular matrix. This paper studies the design, synthesis and characterization of NSAID-peptide conjugates. Attachment of NSAIDs to a diphenylalanine–dilysine (FFKK-OH) peptide sequence generates supramolecular hydrogel forming molecules with antimicrobial and anti-inflammatory properties. NSAID-peptides demonstrate broad-spectrum antimicrobial activity against both Gram-positive and Gram-negative bacteria implicated in a variety of antimicrobial resistant nosocomial infections including Staphylococcus aureus and Pseudomonas aeruginosa. Naproxen-peptides show particular promise, forming biocompatible nanofibrous viscoelastic hydrogel nanosponges composed of β-sheet secondary structures at low concentrations (0.4% w/v). Conjugation of the peptide motif FFKK-OH to naproxen increases selectivity for COX-2 enzyme, implicated in chronic wound scar-tissue formation. Our findings suggest that ultrashort NSAID-peptides have potential use as multifunctional materials for a range of biomedical applications. This includes as topical agents for treatment of chronic wounds, where a profile of persistent inflammation, pain and the presence of infection has been proven to be detrimental to successful wound repair. This work may also serve as a template for the design of future medical device coatings with tailored antimicrobial and anti-inflammatory properties.
 Please wait while we load your content...
Please wait while we load your content...

