DOI:
10.1039/C6RA20242J
(Paper)
RSC Adv., 2016,
6, 108791-108800
Influence of a reagents addition strategy on the Fenton oxidation of rhodamine B: control of the competitive reaction of ·OH†
Received
10th August 2016
, Accepted 11th October 2016
First published on 12th October 2016
Abstract
The Fenton system (Fe2+/H2O2) generates ·OH with a high oxidation potential. However, as reactants themselves, H2O2 and Fe2+ can act as ·OH initiators as well as ·OH scavengers, leading to the need for a high dosage of reactants and increased costs. As a mixing-sensitive reaction, the ·OH-related reaction kinetics (·OH with Fe2+, H2O2, and RhB) was determined from the reaction rates (which were a constant in this work) and stoichiometry, in which the latter could be regulated by an addition strategy of Fenton reagents. This suggests that ·OH competitive reactions could be controlled by applying a macrolevel addition strategy. Herein, the effects of different addition approaches of Fe2+ and H2O2 on ·OH competitive reactions were quantitatively and systematically studied by analyzing the removal of the model pollutant RhB. We found that without stirring, and compared with a one-time addition, once H2O2 or Fe2+ was added in a step-wise pattern (e.g., one drop by one drop, 2 times, or 4 times), a high concentration of H2O2 or Fe2+ existed in a localized place for a longer period, resulting in a lower proportion of ·OH reacting with RhB, which we ascribed to an enhanced reaction between Fe2+, H2O2, and ·OH. However, when H2O2 and Fe2+ were added from two close points without stirring, a larger proportion of ·OH was scavenged by H2O2 and Fe2+; while under stirring, even a one-time addition of H2O2 or Fe2+ could cause severe scavenging of ·OH. The results also revealed a linear relationship between the RhB removal percentage and wavelength blue-shifts. This study showed that microlevel ·OH competitive reactions could be controlled by applying a macrolevel addition strategy of Fenton reagents without the addition of external chemicals. The results suggest this methodology can also offer an approach to lower ·OH invalid consumption by regulating the addition strategy in bigger reactors.
Introduction
The Fenton system (Fe2+ and H2O2), which is one of the advanced oxidation processes (AOPs), generates ·OH to initiate free radical chain reactions and the decomposition of various pollutants.1–4 ·OH is a reactive, nonselective radical, which has a very high standard redox potential (1.9–2.7 V). However, ·OH has a short lifetime, approximately 1 s in the gas phase5 or 10−9 to 10−6 s in the liquid phase.6,7 Therefore, the enhanced generation and effective utilization of ·OH in the Fenton system are key focuses of current research.
As previously reported, the enhanced generation of ·OH in the Fenton system has been well studied experimentally and theoretically.8–10 By the assistance of external energy (i.e., electro-Fenton, photo-Fenton, UV-Fenton, and US-Fenton) and the introduction of specific additives (quinone, hydroxylamine),1,3,11 an improved regeneration of Fe2+ was achieved, which could then react with H2O2 to generate more ·OH. However, the enhanced generation of ·OH cannot guarantee the effective utilization of the generated ·OH. At the same time, the above methods introduced additional chemicals into the system, leading to more expensive running costs.
The reactivity of ·OH is largely dependent on the concentration and availability of hydroxyl radical scavenging compounds.12 Once formed, ·OH reacts rapidly with organic and inorganic substances in solution, with rate constants of 106 to 109 M−1 s−1.13 The key reactions in the Fenton system are shown below.14 Among the reactions involving ·OH, reaction (3) and (4) are the principle ones causing an invalid consumption of ·OH, that is, the desired radical chain propagation is terminated by reaction (3), while weaker radicals (HO2·) are formed by reaction (4). If the side reactions (3) and (4) could be inhibited or weakened, there would be more ·OH available to react with the target pollutants, resulting in an improved overall performance of the Fenton system.
| | |
Fe2+ + H2O2 → Fe3+ + ·OH + OH− (k1 = 76 mol L−1 s−1)1
| (1) |
| | |
Fe3+ + H2O2 → Fe2+ + HO2· + H+ (k2 = 0.02 mol L−1 s−1)1
| (2) |
| | |
Fe2+ + ·OH → Fe3+ + OH− (k3 = 3 × 108 mol L−1 s−1)15
| (3) |
| | |
H2O2 + ·OH → H2O + HO2· (k4 = 2.7 × 107 mol L−1 s−1)16
| (4) |
| | |
2·OH → H2O2 (k5 = 5.2 × 109 mol L−1 s−1)17
| (5) |
| | |
Fe2+ + HO2· → Fe3+ + HO2−
| (7) |
| | |
Fe3+ + HO2· → O2 + Fe2+ + H+
| (8) |
| | |
R· + Fe3+ + OH− → Fe2+ + ROH
| (9) |
| | |
R· + Fe2+ + H+ → Fe3+ + RH
| (10) |
| | |
2R· → RR (in form of dimer)
| (11) |
| | |
R· + O2 → RO2· → RO2− + Fe3+
| (12) |
Based on Shah and Czerlinski's work,18–20 the Fenton reaction is a typical mixing-sensitive reaction. However, we hold the opinion that the essence of this concept is that microlevel ·OH competitive reactions could be influenced by macrolevel stirring and by applying the correct addition strategy of Fenton reagents. Generally, a reaction is determined by the reactants concentration and rate constant. If the reaction parameters (i.e., temperature, pH, and pressure) remained unchanged, then the reactants concentration will be the sole determinant of the reaction. As previous work suggested, the major features of the Fenton system in terms of ·OH production and consumption are believed to be its reagent conditions (i.e., Fe2+, Fe3+, and H2O2).21 However, for a constant reagent concentration, what happens if the reagents addition approach is changed? In this case, a reagent undergoes a diffusion process before it is uniformly dispersed in bulk solution. During this period, the reactant's concentration is changing, thus causing a big difference in the reaction. We thus had the idea that the competitive reactions of ·OH could be regulated by the addition approach taken for adding the reactants.
Some researchers have reported on the effect of the addition approaches on the ·OH scavenging reaction, but they have not systematically analyzed ·OH competitive reactions by H2O2, Fe2+, and the target pollutant. Herein, previous work was understood in terms of ·OH competitive reactions. Many researchers suggested that a high concentration of H2O2 could induce unwanted reactions that consume ·OH.9 To minimize the reaction between H2O2 and ·OH (reaction (4) before), several addition approaches for H2O2 have been evaluated. The results are summarized in Table 1.
Table 1 Results of different addition approaches in the Fenton system by different researchers in the literature
| Compound |
Addition approach |
Findings |
Reference |
| Reactive black 5 |
(1) Pre-dissolved H2O2, followed by one-time or stepped Fe2+ addition |
(1) The effect of the addition strategy was inter-dependent on the mixing intensity level. (2) A higher mixing intensity will enhance the degradation performance when the reagents are added near to each other |
Aris et al. 2010 (ref. 13) |
| (2) Pre-dissolved Fe2+, followed by one-time or stepped H2O2 addition |
| (3) Stepped addition of Fe2+ and H2O2 at two distant points |
| Atrazine |
(1) Total dose of H2O2 was split and added at different times |
(1) The performance of the step-wise Fenton was better. (2) The reaction kinetics of the ATZ decomposition in step-wise Fenton was a two-stage process (a fast decay followed by a stagnant decay) |
Chu et al. 2007 (ref. 22) |
| (2) Total dose of H2O2 was split and added at different quantities |
| Reactive blue 4 |
A central composite experimental design was applied to investigate the effect of five variables (pH, air flow rate, H2O2 flow rate and initial concentrations of Fe(II) and oxalic acid) for the ferrioxalate-assisted solar photo-Fenton process with the continuous addition of H2O2 and air injection |
The degree of dye solution mineralization was enhanced (from 61% to 82%) because the scavenger effect of H2O2 was minimized by the continuous addition of H2O2 |
Montaegudo et al. 2009 (ref. 23) |
| Acrylonitrile-butadiene-styrene |
(1) 4000 mg L−1 H2O2 was added to 2000 mg L−1 Fe2+ solution, which was then agitated with 100 rpm for 2 min and 30 rpm for 158 min |
(1) The step-wise addition of H2O2 and Fe2+ keeps the H2O2 concentration at a relatively low level and reduces the effects of ·OH scavenging |
Popuri et al. 2011 (ref. 24) |
| (2) 4000 mg L−1 H2O2 and 500 mg L−1 Fe2+ were added to the ABS solution, and then Fe2+ solution was added in a step-wise fashion (4 times) for every 40 min |
(2) The batch-wise addition of Fenton's reagent can contribute to the increased removal of DCOD from wastewater |
| (3) 1000 mg L−1 H2O2 and 500 mg L−1 Fe2+ were added to the ABS solution, then H2O2 and Fe2+ solution were added in a step-wise fashion (4 times) for every 40 min |
(3) Agitation has no significant effect on the DCOD removal rate constant or the H2O2 consumption rate constant |
| Azo dye mixtures |
(1) The Fenton's reactants were dosed in a punctual mode |
(1) The dosing strategy influences both the reagent consumption and the biodegradability and toxicity of the effluent |
Garcia et al. 2012 (ref. 25) |
| (2) The Fenton's reactants were dosed continuously |
(2) All the cases showed that feeding the reactor between 40% and 60% of the maximal dose was sufficient to decolorize more than 90% of the mixture of azo dyes |
From Table 1, we can see that adding H2O2 several times can enhance the Fenton performance, which illustrates that once H2O2 is added in a step-wise pattern, the reaction between ·OH and H2O2 will be relieved and more pollutants will be degraded. However, two questions arise: (1) from reaction (3), Fe2+ reacts with ·OH with a rate constant of 3 × 108 mol L−1 s−1, which is ten times larger than the reaction between H2O2 and ·OH (2.7 × 107 mol L−1 s−1), thus radical chain reactions are quickly terminated.21 However, to the best of our knowledge, the inhibition of reaction (3) and the addition approach for Fe2+ solution has not been well studied. (2) As mentioned above, as a fast process, the reactions in the Fenton system are strongly relevant to the diffusion process of the reactants, as well as to stirring. Therefore, a comparative study of stirring or not-stirring cases should be conducted separately to provide more overall information on this aspect.
The objective of the present study was thus to qualitatively investigate different addition approaches of reactants on the competitive reactions in the Fenton system (i.e., ·OH with H2O2, Fe2+, and Fenton's reagents). It should be noted that all the experiments were conducted with the same experimental parameters (i.e., H2O2 concentration, Fe2+ concentration, an initial pH of 3.0). This study offers useful information on the control of the microlevel competitive reactions in the Fenton system by applying a macrolevel addition strategy, thus resulting in a more effective utilization of ·OH.
Materials and methods
2.1 Chemicals
H2O2 (30% solution) and ferrous salt (FeSO4·7H2O) were purchased from Aladdin Chemical Reagent Co. Ltd. Rhodamine B (N,N,N′,N′-tetraethyl rhodamine B, RhB) at 99.5% purity was purchased from Shuang Huan Chemical Reagent Co. Ltd. The structure of RhB is shown below (Fig. 1). Sulfuric acid was used to adjust the solution pH, and dimethyl sulfoxide (DMSO) was used to trap the hydroxyl radicals.26 All the chemicals were used as received.
 |
| | Fig. 1 Rhodamine B chemical structure. | |
2.2 Experimental design
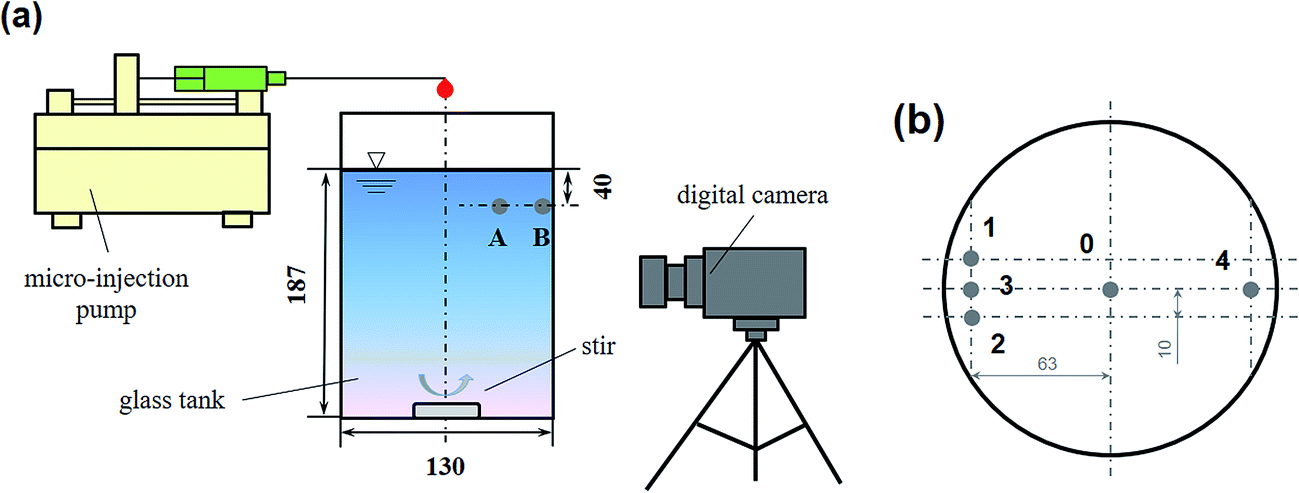
All the experiments were carried out at room temperature (25 ± 1 °C). Aqueous solutions were made using Milli-Q water (18.2 MΩ cm). A 2 L beaker with a PTFE-coated magnetic stirrer was used. The length of the stirrer was 27 mm, which was capable of steadily dispersing the added reagent in a similar pattern (see Fig. S1†). The total volume of the solution was 2040 mL. The initial solution pH was adjusted by sulfuric acid to 3.0. The pH changes in the Fenton system by different addition approaches were monitored, but no obvious change of solution pH was found (see Table S1†). As Jung et al.27 suggested, the lifetime of H2O2 and the overall Fenton reaction are highly affected by the solution pH. Compared with a 2 L solution with an initial pH 3.0, the addition of 20 mL of reagents did not change the solution pH obviously. The experimental system is shown in Fig. 2(a). A digital camera was used to show the diffusion path of a tracer in bulk solution.
 |
| | Fig. 2 (a) Schematic diagram of the beaker system used in this study; (b) the addition position of the reagents (top view). | |
The stock solution of RhB was prepared at 2 mmol L−1 and stored in the dark. The solutions of 20 mL Fe2+ and 20 mL H2O2 were freshly prepared each time to minimize any potential reductions that might otherwise affect the reaction. The reagents were added through a needle (inner diameter: 3 mm) at a selected constant flow rate (for the titration pattern) or added from the center of the beaker directly (for single, 2 times, 4 times patterns). Samples of 1 mL were withdrawn at set intervals (0 min, 2.5 min, 5 min, 7.5 min, 10 min, 12.5 min, 15 min, 17.5 min, 20 min, 30 min), and were diluted to quench the reaction before a UV-Vis analysis was performed. In order to accurately calculate the RhB removal percentage, before sampling, the solution was stirred at 1000 rpm for 2 s and then the direction was reversed at 1000 rpm for another 2 s. In this way, the solution in the beaker could re-stabilize within a short time (ca. 5 s), and this did not influence the following operations.
The addition approaches of Fe2+, H2O2, or Fenton's reagents are listed in Table 2. It should be noted that, in this study, except for run nos A5–A8, all the experiments followed the same parameters: H2O2 at a concentration of 2 mmol L−1, Fe2+ at a concentration of 0.4 mmol L−1, and RhB at a concentration of 0.25 mmol L−1. Runs A1–A4 were designed to study the scavenging effect of Fe2+ on ·OH. Runs B1–B4 were designed to study the scavenging effect of H2O2 on ·OH. Runs a1–a3 and b1–b3 were designed to explore the effect of mixing on the competitive reactions among Fe2+, H2O2, RhB, and ·OH. Runs C1, c2, and c3 were designed to study the co-scavenging effect of Fe2+ and H2O2 on ·OH. The addition position is shown in Fig. 2(b). For runs A1–A8, a1–a3, B1–B4, and b1–b3, H2O2 or Fe2+ were added to the bulk solution from position 0. For run C1, Fe2+ and H2O2 were added from positions 1 and 2. For runs c2 and c3, Fe2+ and H2O2 were added from positions 3 and 4.
Table 2 Lists of the addition approaches applied in this work
| Addition approach |
Run no. |
| Total Fe2+ solution was split and added at 0 min and 10 min. Compared with runs A1–A4, runs A5–A8 involved an increased concentration of Fenton reagents and RhB (H2O2: 5 mmol L−1; Fe2+: 1 mmol L−1; RhB: 0.3 mmol L−1). |
| Pre-dissolved H2O2, followed by the addition of Fe2+ |
Without stirring |
Adding Fe2+ at 0 min |
A1 |
| Adding Fe2+ at 0 and 10 mina |
A2 |
| Adding Fe2+ at 0, 5, 10, 15 min |
A3 |
| Step addition within 30 min |
A4 |
| Adding Fe2+ at 0 minb |
A5 |
| Adding Fe2+ at 0 and 10 minb |
A6 |
| Adding Fe2+ at 0, 5, 10, 15 minb |
A7 |
| Step addition within 30 minb |
A8 |
| With stirring |
Adding Fe2+ at 0 min, 300 rpm |
a1 |
| Adding Fe2+ at 0 min, 1500 rpm |
a2 |
| Step addition within 30 min, 1500 rpm |
a3 |
| Pre-dissolved Fe2+, followed by the addition of H2O2 |
Without stirring |
Adding H2O2 at 0 min |
B1 |
| Adding H2O2 at 0 and 10 min |
B2 |
| Adding H2O2 at 0, 5, 10, 15 min |
B3 |
| Step addition within 30 min |
B4 |
| With stirring |
Adding H2O2 at 0 min, 300 rpm |
b1 |
| Adding H2O2 at 0 min, 1500 rpm |
b2 |
| Step addition within 30 min, 1500 rpm |
b3 |
| Step-wise addition of H2O2 and Fe2+ |
Step addition of H2O2 and Fe2+ at two close points, without stirring |
C1 |
| Step addition of H2O2 and Fe2+ at two distant points within 10 min, 300 rpm |
c2 |
| Step addition of H2O2 and Fe2+ at two distant points within 30 min, 300 rpm |
c3 |
2.3 Analytical methods
The results of the competitive reactions among Fe2+, H2O2, RhB, and ·OH were evaluated based on the RhB removal efficiency. The concentration of RhB was analyzed based on the absorbance peak at wavelength 554 nm (ref. 28) using a UV-Vis spectrometer (Varian Carry 300, USA). The efficiency of RhB removal was calculated with the following equation (eqn (13)), where C0 is the calculated initial concentration of RhB and Ct is the final measured concentration of RhB solution. Since the disappearance of RhB is a result of oxidation by ·OH28 (see Fig. S2†), any reaction that scavenges ·OH will significantly affect the rate of RhB removal. Based on the experimental design in Table 2, the key feature of the addition approaches tested in this work is that H2O2 or Fe2+ reagent undergoes diffusion within 30 min, thus making it difficult to accurately detect the Fe2+ or H2O2 concentration. The sample solution at a specific location in a 2 L beaker cannot represent the concentration at other places since Fe2+ or H2O2 are not distributed uniformly in the solution. The change of Fe2+ concentration in a typical well-mixed Fenton system is shown in Fig. S3.†29| |
 | (13) |
TOC was determined using a TOC-5050 Shimadzu analyzer (standard deviation < 0.2 mg L−1). The oxidation and reduction potentials of the Fenton system under various conditions were measured with an ORP composite electrode (E501, China),30 while the solution pH value was measured and monitored with a pH-meter (E201-C, China).
Results and discussion
3.1 Visualization of the mixing process using a tracer in bulk solution
The design of different addition approaches aimed to create different competitive situations for ·OH-related reactions. Herein, concentrated methylene blue (blue in solution) and rhodamine B (red in solution) were used as tracers to show the mixing process under various addition approaches. The results are shown in Fig. 3.
 |
| | Fig. 3 Mixing process of a tracer in bulk solution following addition by different addition approaches: (a) adding one drop of tracer without stirring; (b) one-time addition of 20 mL of tracer without stirring; (c) one-time addition of 20 mL of tracer with stirring at 300 rpm; (d) one-time addition of 20 mL of tracer with stirring at 1500 rpm; (e) adding one drop of tracer with stirring at 1500 rpm; (f) adding two kinds of tracers from distant positions with stirring at 300 rpm (one drop, respectively). | |
As (a) and (b) suggest, tracer was not well dispersed without stirring, even after 115 s for case (a). Compared with (b), it is obvious that once the tracer was added by one drop, it is difficult to be well dispersed in bulk solution, especially at room temperature, which implied that a relatively high concentration of tracer could be maintained for a longer time. For (c) and (d), the characteristic times were 15 s and 8 s, respectively, which showed the tracer was dispersed uniformly in a shorter time. Case (e) showed that under stirring cases, one drop of tracer concentrated at the center of the beaker, then diffused to the bulk solution. As (f) shows, when two tracers are added from distant points (positions 3 and 4, as illustrated in Fig. 2(b)), they encounter each other in a relatively low concentration. It is therefore reasonable to assume that the mixing process is also applicable to the various addition approaches mentioned in Table 2, in which Fenton reagents could be referred to as the tracer.
3.2 Pre-dissolving H2O2, followed by different addition approaches for Fe2+
As seen in reactions (1) and (3), Fe2+ can act as an ·OH initiator as well as an ·OH scavenger. Many researchers have focused on the addition approach for H2O2, but only Popuri et al.24 have studied the addition approach for Fe2+. Herein, the step-wise addition of Fe2+ was studied from a viewpoint of competitive reactions (A1–A4 and A5–A8).
Ideally, Fe2+ reacts with H2O2 when they are dispersed uniformly in bulk solution. However, once Fe2+ is added to an H2O2 bulk solution in a step-wise pattern, the mixing process of Fe2+ influences the reaction, thus causing a change in the ·OH competitive reactions.
To evaluate the reaction between Fe2+ and ·OH, several addition approaches were designed. All the experiments in this part (A1–A4) were conducted without stirring. As Section 3.1 suggests, when Fe2+ is added one drop by one drop within 30 min, each drop of Fe2+ will take a long time to disperse, and so a high concentration Fe2+ will remain for a long time. Therefore, the temporary localized Fe2+ could result in a severe reaction between Fe2+ and ·OH (for a more detailed explanation, see ESI: Calculation 1†). However, when 20 mL of Fe2+ solution is added on a one-time basis, all the Fe2+ will be dispersed in a shorter time, far less than in the case of A1, whereby the reaction between Fe2+ and ·OH will be alleviated. By this design, a different consumption of ·OH can possibly be achieved. The results are shown in Fig. 4(a), while the results for the increased concentration of Fenton reagents and RhB are shown in Fig. 4(b).
 |
| | Fig. 4 (a) The degradation of 0.25 mM RhB with an initial pH of 3.0 and 2 mM H2O2 by different addition approaches for Fe2+. (b) The effect of an increased concentration of Fenton reagents and RhB on RhB degradation by different addition approaches for Fe2+. Conditions: [H2O2] = 5 mM; [Fe2+] = 1 mM; [RhB] = 0.3 mM; initial pH 3.0; T = 25 °C. | |
Fig. 4(a) successfully confirmed our hypothesis. A1 achieved the highest removal percentage, 89.4%, while A4 showed the lowest removal effect, 52.42%. This implied that once Fe2+ was added one drop by one drop, more ·OH was consumed by the surrounding Fe2+ via reaction (3), causing a lower proportion of ·OH to be available to react with RhB, as shown in Fig. 5. In this case, the reaction stoichiometry was changed, resulting in a localized Fe2+ concentration much higher than 0.4 mM (for a more detailed explanation, see ESI: Calculation 1†), which lasts for several seconds (more than 115 s, as shown in Fig. 3(a)), resulting in a strong reaction between Fe2+ and ·OH. Meanwhile, we also found that when the concentration of reactants was increased, nearly all the RhB molecules could be degraded by ·OH, which cannot reflect the reaction between Fe2+ and ·OH, as Fig. 4(b) shows. Fig. 6 also shows that during the degradation of RhB, some intermediates are formed, leading to an increase in the absorption from 300–450 nm.27 Herein, the influence of the intermediates on the ·OH competition reactions was not included.
 |
| | Fig. 5 Competitive reactions among Fe2+, H2O2, RhB, and ·OH once Fe2+ was added on a one drop by one drop basis. | |
 |
| | Fig. 6 The temporal UV-Vis spectra of RhB during its degradation of 30 min by different addition approaches for Fe2+: (a) one-time addition of Fe2+ at 0 min; (b) four-times addition of Fe2+ at 0, 5, 10, 15 min (note, the sample solution was diluted 25-fold). | |
3.3 Pre-dissolved Fe2+, followed by different addition approaches for H2O2
From reactions (1) and (4), it can be seen that H2O2 acts as an ·OH initiator as well as an ·OH scavenger. Research into an H2O2 addition approach has been well studied by several researchers.12,17,22 However, most of the research was conducted with stirring. As is known, mixing could exert a huge influence on the reagents dispersal, especially for mixing-sensitive reactions.18,19 This is therefore a key topic of this study and consequently, several experiments were designed to study the addition approach for H2O2 on the reaction between H2O2 and ·OH, on the basis of an analysis of RhB removal.
All the experiments in this part (B1–B4) were conducted without stirring. The same as with A1–A4, when H2O2 solution was added continuously within 30 min, it was difficult for each drop of H2O2 to disperse in bulk solution, and excessive H2O2 molecules depleted the valuable ·OH to form HO2·. Since HO2· was less reactive than ·OH,31 the removal of RhB was therefore reduced. The one-time addition of H2O2 followed the same mixing process introduced above. The results are shown in Fig. 7. The one-time addition of H2O2, in which H2O2 could disperse more easily compared with in a stepped addition, achieved the highest removal percentage of RhB. Besides, compared with Fig. 4, the removal of RhB by the step-wise addition of H2O2 did not show any obvious “step decrease”.
 |
| | Fig. 7 The degradation of 0.25 mM RhB with the initial pH of 3.0 and 0.4 mM Fe2+ by different addition approaches for H2O2. | |
Fig. 8(a) shows the results of A1 and B1, while Fig. 8(b) shows the oxidation–reduction potential of two addition processes (ORP is used as an indicator of the oxidation–reduction potential of the solution and mainly represents the Fe3+ level, see ESI: Fig. S4†). These results implied that whether adding H2O2 to Fe2+ or the other way round in terms of Fe2+ to H2O2 does not show a big difference, which suggests that in these two cases, the scavenging effect of H2O2 to ·OH and Fe2+ to ·OH are generally the same.
 |
| | Fig. 8 Comparison of the one-time addition of Fe2+ to H2O2 and the one-time addition of H2O2 to Fe2+ without stirring. (a) The degradation of 0.25 mM RhB, using data from Fig. 1 and 4. (b) Profile of ORP change. | |
3.4 Step-wise addition of H2O2 and Fe2+
In this part, we designed a series of experiments that could cause a bad condition for ·OH once generated. For C1, H2O2 and Fe2+ were continuously added within 30 min at two close points (positions 1 and 2). In this way, a high concentration of ·OH was generated in a localized place, together with an environment of a high concentration of H2O2 and Fe2+, which could severely consume ·OH.13 For c2 and c3, H2O2 and Fe2+ were added at two distant points (c2, 300 rpm, adding within 10 min; c3, 1500 rpm, adding within 30 min; positions 3 and 4), which could cause a better condition for ·OH utilization. The results are shown in Fig. 9.
 |
| | Fig. 9 Comparison of the step addition of Fe2+ and H2O2 under different conditions. (B1) One-time addition of H2O2 to Fe2+, using data from Fig. 4; (C1) step addition of H2O2 and Fe2+ at two close points, without stirring; (c2) step addition of H2O2 and Fe2+ at two distant points within 10 min, 300 rpm; (c3) step addition of H2O2 and Fe2+ at two distant points within 30 min, 300 rpm. | |
C1 showed the lowest removal percentage of RhB at 30 min, which was in accordance with our hypothesis above. Besides, as Fujishima et al. reported,32 ·OH has a short lifetime (ca. 10 μs), therefore a localized high concentration of ·OH cannot diffuse to a remote place and contribute to RhB degradation. In c3, although H2O2 and Fe2+ were added at two distant points, C1 and c3 did not show a big difference, which was ascribed to the influence of mixing on ·OH utilization. This phenomenon is still unknown up to now. For c2, once H2O2 was added within a shorter time, i.e., 10 min, a more efficient degradation of RhB could be achieved. This was attributed to the enhanced generation of ·OH compared with c3.
3.5 Influence of mixing on RhB degradation
If the reagents cannot disperse effectively, a high concentration of ·OH could be localized in central areas within the beaker. Such a condition causes a severe invalid reaction between ·OH and Fe2+ or H2O2. Whether ·OH can diffuse to remote places and oxidize pollutants is an interesting question. Runs a1–a3 and b1–b3 were designed to study the effect of mixing on competitive reactions and RhB removal. The results are shown in Fig. 10.
 |
| | Fig. 10 The influence of mixing on RhB degradation. (a) Pre-dissolved H2O2, followed by the addition of Fe2+; (b) pre-dissolved Fe2+, followed by the addition of H2O2. | |
As Fig. 10(a) and (b) show, a1–a3 and b1–b3 followed a similar rule. The one-time addition of Fe2+ or H2O2 with 300 rpm or 1500 rpm stirring caused a lower removal of RhB (89.4% for A1, 68.3% for a1, 64.2% for a2). Once Fe2+ or H2O2 was added continuously with stirring at 1500 rpm, the degradation of RhB occurred gradually. The curves of a1, a2, and A1 (b1, b2, and B2) are quite different due to the stirring. However, it should be noted that the removal percentage of RhB at 2.5 min did not show much difference (53.2% for A1, 46.2% for a1, 45.9% for a2). This illustrated that stirring possibly cannot facilitate the diffusion of ·OH to remote places, and may even exert reverse effects on RhB oxidation. Although stirring with 1500 rpm mixed the solution efficiently, ·OH-forming reactions are fast and ·OH has a short life (ca. 10 μs) in the liquid phase, thus meaning it functions at a localized position.
This result was reproduced in another repeated run. Based on the diffusion time discussed in Section 3.1, from 0 min to 2.5 min, stirring and non-stirring cases function on the Fenton reaction by impacting the diffusion process. From 2.5 min to 30 min, the two systems are the same, except that the high mixing level leads to an increase in DO concentration in a1 and a2. Therefore, the difference on RhB removal could be ascribed to the drawn-in O2 on the oxidants in the Fenton system, which was also suggested in the work of Huston,33 Hislop,34 and Aris.35
3.6 Comparison of the blue-shift of RhB solution by applying different addition approaches
Zhuang et al.36 reported that in the TiO2 photocatalysis system, surface ·OH and bulk ·OH could attack different parts of methylene blue molecules, causing a decrease or blue-shift of the major absorption band. Surprisingly, in the homogeneous Fenton system, we detected a huge difference in the wavelength shifts among different runs. Fig. 11 shows the results of the wavelength shifts of the major absorption band under pre-dissolved H2O2, followed by Fe2+ addition. Fig. 12 is the results of the wavelength shifts of the major absorption band under pre-dissolved Fe2+, followed by H2O2 addition.
 |
| | Fig. 11 Wavelength shifts of the major absorption band under pre-dissolved H2O2, followed by Fe2+ addition: (a) without stirring; (b) with stirring; (c) increased concentration of reactants. Conditions for (c): [H2O2] = 5 mM; [Fe2+] = 1 mM; [RhB] = 0.3 mM; initial pH 3.0; T = 25 °C. | |
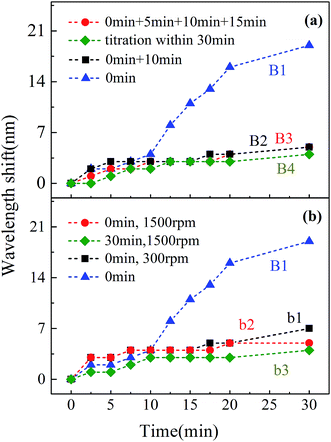 |
| | Fig. 12 Wavelength shifts of the major absorption band under pre-dissolved Fe2+, followed by H2O2 addition: (a) without stirring; (b) with stirring. | |
As Fig. 11 shows, in A1–A4 and a1–a3, the one-time addition of Fe2+ showed the maximum wavelength shift with or without stirring. For B1–B4 and b1–b3, the one-time addition of H2O2 showed the same rule as for Fig. 11. For A5–A8, the wavelength shifts could achieve a high value. Concurrently, nearly 100% degradation was achieved, as shown in Fig. 4. Furthermore, it is obvious that other addition approaches achieved a smaller value at 30 min, less than 6 nm. In order to further explore the wavelength shifts during 30 min, data for the removal rate and wavelength shifts are summarized and shown in Fig. 13.
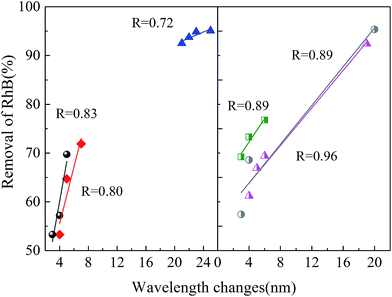 |
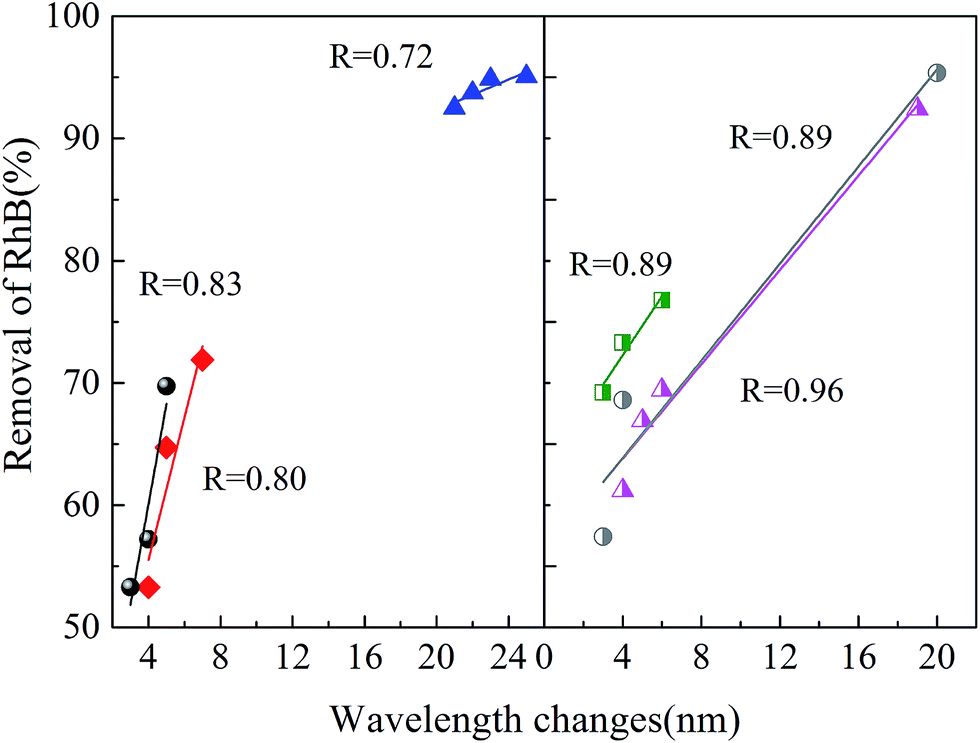
| | Fig. 13 Relationship between RhB removal percentage and the wavelength shifts. | |
In all the experiments, the removal percentage of RhB showed a linear relationship with the wavelength shifts. As could be seen from the UV-Vis spectra, a gradual decrease in the major absorption band means the removal of the chromophore group in the RhB molecule structure, while the shifts of the major absorption band mean the removal of the auxochrome group (N-deethylation). Therefore, the linear relationship of the two parameters means that the removal of the chromophore group of RhB and N-ethyl occurred simultaneously, and the removal of N-ethyl made the removal of chromophore group easier. The results of the wavelength shifts were in accordance with our former conclusion, in which a higher wavelength shift means more ·OH reacting with RhB molecules.
It should be noted that compared with the work of Zhuang et al.,36 the maximum wavelength shift in this work is 27 nm, smaller than their 34 nm. This is because the degradation of RhB is a step-by-step deethylation to give several intermediate products, such an N,N,N′-triethyl rhodamine (TER, 539 nm), N,N′-diethyl rhodamine (DER, 522 nm), N-ethyl rhodamine (MER, 510 nm), and rhodamine at 498 nm.37 Among all the runs, the maximum TOC removal was 14.3% (see ESI Fig. S5†), which showed no complete mineralization of RhB.
Conclusions
In this study, the effects of the addition approach of the Fenton reagents on ·OH competitive reactions were quantitatively studied on the basis of RhB removal. A tracer was used to show the feasibility of our experimental design. The results showed that: without stirring, a step-wise addition of H2O2 or Fe2+ (one drop by one drop, 2 times, 4 times) showed an undesirable mixing process, resulting in a severe scavenging effect for ·OH; whereas once H2O2 and Fe2+ were added from two close points without stirring, a larger proportion of ·OH was scavenged by H2O2 and Fe2+. Under stirring cases, even the one-time addition of H2O2 or Fe2+ could cause a severe scavenging effect for ·OH. The data also showed a linear relationship between the RhB removal percentage and wavelength blue-shifts. These results seem to support our aforementioned assertion with regard to the occurrence of the localized generation of ·OH causing the scavenging of ·OH. This work systematically presents ·OH competitive reactions with H2O2, Fe2+, and RhB by different addition approaches of Fenton reagents. The results showed that microlevel competitive reactions could be controlled by applying a macrolevel addition strategy of reactants. The results can also offer insights to lower the ·OH invalid consumption by regulating the addition strategy in a bigger reactor.
Acknowledgements
This work was supported by the National Natural Science Foundation of China under Grant No. 91434134 and No. 51421063.
References
- J. H. Ma, W. J. Song and C. C. Chen, Environ. Sci. Technol., 2005, 39, 5810 CrossRef CAS PubMed.
- J. J. Pignatello, E. Oliveros and A. Mackay, Environ. Sci. Technol., 2006, 36, 1 CrossRef CAS.
- L. W. Chen, J. Ma and X. C. Li, Environ. Sci. Technol., 2011, 45, 3925 CrossRef CAS PubMed.
- Z. M. Qiang, J. H. Chang and C. P. Huang, Water Res., 2003, 37, 1308 CrossRef CAS PubMed.
- I. S. A. Isaksen and S. B. Dalsøren, Science, 2011, 331, 38 CrossRef CAS PubMed.
- Y. Murakami, K. Endo, I. Ohta, A. Y. Nosaka and Y. Nosaka, J. Phys. Chem. C, 2007, 111, 11339 CAS.
- J. M. Monteagudo, A. Durán, S. Martín and M. Aguirre, Appl. Catal., B, 2009, 89, 510 CrossRef CAS.
- Y. Y. Wu, S. Q. Zhou, F. H. Qin, K. Zheng and X. Y. Ye, J. Hazard. Mater., 2010, 179, 533 CrossRef CAS PubMed.
- J. D. Laat and H. Gallard, Environ. Sci. Technol., 1999, 33, 2726 CrossRef.
- Y. X. Du, M. H. Zhou and L. C. Lei, Water Res., 2007, 41, 1121 CrossRef CAS PubMed.
- L. S. Luo, Y. Y. Yao, F. Gong, Z. F. Huang, W. Y. Lu, W. X. Chen and L. Zhang, RSC Adv., 2016, 6, 47221 Search PubMed.
- J. E. Donham, E. J. Rosenfeld and K. R. Wigginton, Environ. Sci.: Processes Impacts, 2014, 16, 764 CAS.
- A. Aris and P. N. Sharratt, Environ. Technol., 2004, 25, 601 CrossRef CAS PubMed.
- B. S. Lane and K. Burgess, Chem. Rev., 2003, 103, 2457 CrossRef CAS PubMed.
- H. Christensen, K. Sehested and H. Corfitzen, J. Phys. Chem., 1982, 86, 1588 CrossRef CAS.
- Z. Stuglik and Z. P. Zagorski, Radiat. Phys. Chem., 1981, 17, 229 CrossRef CAS.
- K. Sehested, E. Bjergbakke and O. L. Rasmussen, J. Chem. Phys., 1969, 51, 3159 CrossRef CAS.
- S. I. A. Shah, L. W. Kostiuk and S. M. Kresta, Int. J. Chem. Eng., 2012, 2012, 1, DOI:10.1155/2012/654321.
- S. I. A. Shah, L. W. Kostiuk and S. M. Kresta, Int. J. Chem. Eng., 2012, 2012, 1, DOI:10.1155/2012/750162.
- G. Czerlinski, R. Levin and T. Ypma, Int. J. Chem. Kinet., 2003, 35, 484 CrossRef CAS.
- E. Neyens and J. Baeyens, J. Hazard. Mater., 2003, 98, 33 CrossRef CAS PubMed.
- W. Chu, K. H. Chan and C. Y. Kwan, Chemosphere, 2007, 67, 755 CrossRef CAS PubMed.
- J. M. Monteagudo, A. Durán and S. Martín, Chem. Eng. J., 2010, 162, 702 CrossRef CAS.
- S. R. Popuri, C. Y. Chang and J. Xu, Desalination, 2011, 277, 141 CrossRef CAS.
- D. P. Garcia and G. Buitrón, J. Hazard. Mater., 2012, 218, 293 CrossRef PubMed.
- H. Q. Zhao, J. H. Gao and W. Zhou, Anal. Methods, 2015, 7, 5447 RSC.
- Y. S. Jung, W. T. Lim and J. Y. Park, Environ. Technol., 2009, 30, 183 CrossRef CAS PubMed.
- M. F. Hou, L. Liao and W. D. Zhang, Chemosphere, 2011, 83, 1279 CrossRef CAS PubMed.
- W. Zhou, H. Q. Zhao and J. H. Gao, Environ. Technol., 2016 DOI:10.1080/09593330.2016.1240241.
- F. Chen, W. H. Ma and J. J. He, J. Phys. Chem. A, 2002, 106, 9485 CrossRef CAS.
- K. H. Chan and W. Chu, Environ. Technol., 2003, 24, 703 CrossRef CAS PubMed.
- A. Fujishima, X. T. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515 CrossRef CAS.
- P. L. Huston and J. J. Pignatello, Environ. Sci. Technol., 1996, 30, 3457 CrossRef CAS.
- K. A. Hislop and J. R. Bolton, Environ. Sci. Technol., 1999, 33, 3119 CrossRef CAS.
- A. Aris and P. N. Sharratt, Environ. Technol., 2006, 27, 1153 CrossRef CAS PubMed.
- J. D. Zhuang, W. X. Dai and Q. F. Tian, Langmuir, 2010, 26, 9686 CrossRef CAS PubMed.
- T. Watanabe, T. Takizawa and K. Honda, J. Phys. Chem., 1977, 81, 1845 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra20242j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Haiqian Zhao
a,
Haiqian Zhao