
Ring opening polymerization of ε-caprolactone in the presence of wet β-cyclodextrin: effect of the operative pressure and of water molecules in the β-cyclodextrin cavity
Alessandro Galia*a,
Onofrio Scialdonea,
Tiziana Spanòa,
Maria Grazia Valentia,
Bruno Grignarda,
Philippe Lecomteb,
Eric Monflierc,
Sébastien Tilloyc and
Cyril Rousseauc
aDipartimento Ingegneria Chimica Gestionale Informatica Meccanica, Università di Palermo, Viale Delle Scienze Ed.6, 90128 Palermo, Italy. E-mail: alessandro.galia@unipa.it; Fax: +39-091-23860841; Tel: +39-091-23863758
bCenter for Education and Research on Macromolecules (CERM), University of Liege, B6a Sart-Tilman, B-4000 Liege, Belgium
cUniv. Artois, CNRS, Centrale Lille, ENSCL, Univ. Lille, Unité de Catalyse et de Chimie Du Solide (UCCS), UMR 8181, F-62300 Lens, France
First published on 12th September 2016
Abstract
The ring opening polymerization (ROP) of ε-caprolactone (CL) in the presence of β-cyclodextrin (β-CD) was performed in batch reactors both at room pressure and with the reaction system pressurized with CO2, N2 or Ar. Significant enhancements of the polymerization rate was observed when the ROP was carried out with wet β-CD under pressure. For example, after 24 hours at 120 °C with a β-CD/CL molar ratio of about 1/100, the monomer conversion increased from 4 to 98–99% when the pressure was changed from 0.1 to 12.5–13.0 MPa independent of the nature of the compressing gas. MALDI-TOF analyses indicated that a major fraction of polymer chains obtained in pressurized systems was initiated by water molecules. The collected results suggest that at 12–13 MPa wet β-CD can catalyse both the ring opening of ε-caprolactone and the polymerization of the obtained linear species and that high energy water molecules located inside the cavity of the cyclic oligosaccharide must play a role in initiating the polymerization. The trend of number average molecular weight and the results of MALDI-TOF analyses obtained in polymerizations performed for long reaction times and in a hydrolysis test of commercial poly(ε-caprolactone) indicate that wet β-CD can work as an artificial lipase enzyme under the adopted conditions.
1. Introduction
Aliphatic polyesters are biodegradable polymers that are the object of steadily increasing interest owing to their potential applications in packaging, biomedical devices and drug delivery systems.1–3 Additionally they can be prepared from renewable monomers thus further increasing the sustainability of their utilization.4In order to overcome thermodynamic constraints related to the accumulation of the condensate in step growth polymerization the best approach to synthesise polyesters is to perform ring opening polymerization (ROP) of cyclic aliphatic esters in the presence of a suitable catalyst. Metal alkoxides are the most widely adopted catalysts to activate ROP of cyclic esters.5 These compounds generally operate by a coordination–insertion mechanism and they are generated by the reaction of suitable precursors with an alcohol molecule that works as initiator of the polymerization thus being incorporated as chain end group in the synthesized macromolecule (Scheme 1).
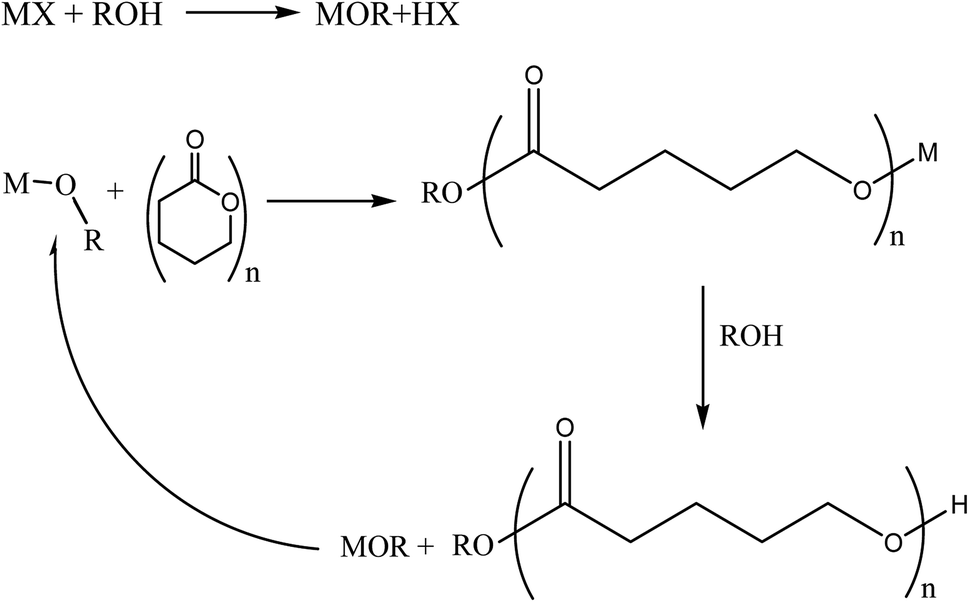 | ||
| Scheme 1 Schematic description of coordination–insertion mechanism of metal alkoxide catalysed ROP of cyclic aliphatic esters. M = transition or rare earth metal, X = supporting ligand, OR alkoxide group. | ||
In the case of biomedical applications it would be highly interesting to activate the polymerization with non metal catalysts to avoid contamination of the macromolecular matrix that could adversely affect its biocompatibility.
To this regard enzymes have been tested as catalysts with interesting results but their high cost makes difficult the transfer of this synthetic approach from the laboratory to the industrial plant.6–8
Naturally occurring β-cyclodextrins (β-CD) are truncated cone-shaped molecules with a tapered cavity 7.9 Å deep with top and bottom diameters of 6.0 and 6.5 Å. Together with their α- and γ-homologues, they are well known to possess a hydrophobic cavity capable of forming inclusion complexes with many organic molecules which are otherwise poorly soluble in water (Scheme 2). This property is commonly attributed to van der Waals and hydrophobic interactions between the host β-CD cavity and the guest molecule even if the role of hydrogen bonding and steric effects cannot be ruled out.9
 | ||
| Scheme 2 Schematic representation of native cyclodextrins. | ||
Such molecular recognition capability joined to their site-specific chemical reactivity towards selected reactions that can be enhanced by proper functionalization of the CD make them interesting examples of artificial enzymes or chemzymes.10–12
It has been shown that native cyclodextrins can be used as co-initiators of the ROP of cyclic esters.13,14 Moreover, Harada and coworkers found that the ROP of aliphatic cyclic esters can be activated by native cyclodextrins leading to the formation of macromolecular chains in which the cyclic oligosaccharides are incorporated as chain ends acting then as initiators.15–18 Polymerizations were performed in the absence of any solvent or co-catalyst, under strictly anhydrous conditions and with high molar ratio of CD to monomer. The polymerization rate was found to be dependent on the good steric matching between the cyclic ester and the CD cavity thus suggesting that the formation of an inclusion complex is essential for the activation of the ROP. As further support of this hypothesis it can be considered that when the polymerization of δ-valerolactone (δ-VL) was carried out adding to the bulk monomer the β-CD-adamantane inclusion complex, that is well known to have the basket firmly occupied owing to the high constant of its inclusion equilibrium, the rate of polymerization was strongly depressed.16 By functionalization of the 2-hydroxyl of α-CD with cinnamoyl group, deactivation of the polymerization can be achieved by photoinduced cis to trans isomerization of the cinnamoyl group.19 Harada and coworkers have also shown that the terminal β-CD units of poly(δ-VL), both single or arranged in supramolecular nanospheres, are active polymerization sites that allows the operator to extend the polymer chain provided that poly-pseudo-rotaxanes are formed.17,20,21 In these systems, tethered cyclodextrins impart a linear conformation to the polyester chain so that the basket of the terminal CD remains accessible to the cyclic monomer that will be included in the macromolecular chain. On the basis of these information dimeric cyclodextrins have been synthesized using suitable bifunctional molecules that behave as molecular spacers between the two cyclic oligosaccharides. Such CD dimers exhibit a behavior similar to polymerases for the polymerization of δ-VL without co-catalysts or solvents.22
These results are very interesting in the perspective of synthesizing polyesters for biomedical applications from renewable monomers in the absence of any metal catalysts and their transfer to applications would make easier if high molar ratio of monomer to CD could be used and if polymerization time could be decreased particularly in the case of larger cyclic monomers. For example in the case of the ROP polymerization of ε-caprolactone (CL) performed with β-CD using a molar ratio CL/β-CD of 5, polymer yields of 15% were obtained at 100 °C after 72 hours.16
It must be precised that native cyclodextrins are not soluble in the bulk monomers.18 In this case the reaction rate could be also dependent on the rate of mass transfer of the monomer to the active site that is hypothesized to be the cavity of the CD attached at one end of the growing polymer chain. The kinetics of this mass transfer process could be decreased by the enhancement of the viscosity of the polymerization medium when the polymer chains accumulate and increase their length.
Supercritical carbon dioxide (scCO2) has a relatively high solubility in poly(ε-caprolactone) (PCL)23 and, as a consequence of its dissolution in the matrix, depression of the polymer melting point24 and of the local viscosity of the molten polymer25 have been observed. On the other hand, at high enough density, scCO2 can solubilize significant concentrations of CL.26
On the basis of aforementioned background we started an investigation of the ROP of CL in the presence of scCO2, at pressure low enough that it behaves just as an expanding agent of the molten polymer phase, to study its effect on the performances of the polymerization. As a comparison, also non-swelling permanent gases such as Ar and N2 were used as pressurizing agent of the polymerization medium to decouple any effect of pressure and plasticization on the performances of the polymerization. Experiments were performed at 393–423 K and pressure lower than 13 MPa, at these operative conditions the density of CO2, as estimated by the Span & Wagner equation of state,27 is too low to exhibit significant solvent power both for the monomer26 and for the native cyclodextrin.28 During this study we have found that pressure can significantly accelerate the rate of the polymerization of ε-CL provided that wet β-CD is used as catalyst and, under adopted conditions, only a minor fraction of the polyester chains is terminated by the CD, the main end group being constituted by hydroxyl and carboxyl groups arising from water initiation. We have decided to investigate in detail this unexpected behavior and the results that we have obtained are here reported.
2. Experimental
2.1 Materials
ε-Caprolactone with a purity of 99% was supplied by Alfa Aesar. It was used both as received and desiccated over molecular sieves 4 Å (Sigma Aldrich) according to a procedure reported in the literature.21 β-Cyclodextrin with a purity higher than 97% was supplied by Sigma Aldrich. In some experiments it was dried at 80 °C under vacuum for 5 hours immediately before its utilization. Water Sigma Aldrich HPLC grade was added to the reaction system in some of the experimental tests. CO2 (purity 99.998%), N2 (purity 99.9990%) and Ar (purity 99.9990%) were all supplied by Air Liquide. Commercial PCL was purchased from Aldrich. Its number average degree of polymerization was estimated by 1H-NMR spectroscopy in CDCl3.2.2 Reaction systems and procedures
Reactions performed at room pressure were carried out in glass Schlenk tubes equipped with a valve which allowed us to evacuate it and to fill it with an inert gas. The reaction temperature was controlled by insertion of the glass reactor in a silicon oil bath placed on a hot plate magnetic stirrer (Velp Scientifica Arex) equipped with a digital thermoregulator which uses Fuzzy logic technology.Reactions under pressure were carried out in an AISI 316 reactor, having a total volume of 27 mL. It consists of a cylindrical body and a threaded head. A Parker valve and a Barksdale UPA 3 pressure transducer were screwed on the head in which there is also a well for the insertion of a type K thermocouple. The reactor was heated to the reaction temperature by an electric heating mantle driven by an Eurotherm PID controller.
Weighed amounts of β-CD and CL were loaded in the glass tube or in the reactor. Selected tests were performed in the absence of β-CD adding water to the monomer. Prior to these tests, the reactor was cleaned loading it with a tetrahydrofuran (THF)/carbon dioxide mixture 50/50 w/w in such amount to reach a nominal density of 0.9 g mL−1 and heating the system to 373 K. The loaded reactor was treated for 1 hour in its standard position and for 1 hour inverting its orientation to clean also the ducts in the threaded head of the vessel.
When the monomer had to be dried prior its utilization, the CL was treated with molecular sieves 4 Å activated at 473 K under N2 stream for 24 hours. After cooling under nitrogen stream a suitable amount of molecular sieves (20% w/w with respect to the mass of CL to be treated) were added to the monomer in a glass bottle sealed by Suba Seal rubber septum. A treatment time of 72 hours was waited for before the monomer was considered ready for the utilization29 and it was transferred to the reactor by a syringe under nitrogen atmosphere weighing its amount by a Sartorius LP8200S scale (precision ± 0.01 g). The amount of β-CD loaded in the reactor was weighed by an electronic scale Sartorius CP 225D (precision 0.01 mg). After addition of these two components the reactor was closed and atmospheric oxygen was carefully removed by several low pressure chromatographic nitrogen washes in the case of the glass reactors or by connection to an Edwards vacuum pump for 15 minutes in the case of the AISI 316 reactors. During these treatments, the reaction mixture is stirred by a magnetic stir bar to assist desorption of the dissolved oxygen. After this treatment the reactor is pressurized with CO2, N2 or Ar at the desired pressure and heated at the polymerization temperature (usually 393 K).
At the end of the polymerization time the reactor is cooled by submerging it in a bath of ice and water and the polymerization mixture is collected and stored under dark and at low temperature (255 K).
2.3 Analytical techniques
The product collected at the end of the experiments was analyzed using 1H-NMR analyses with a Bruker AN 400 spectrometer, using deuterated chloroform as the solvent, at 298 K. Chemical shifts were given in parts per million with tetramethylsilane as an internal reference. In Fig. 1 are reported two spectra respectively of the monomer and the polymer, the conversion X of CL and the number average degree of polymerization DPn of the obtained polymer were estimated through the following equations:| X = Ad/(Aε+Ad) | (1) |
| DPn = Ad/Af | (2) |
 | ||
| Fig. 1 Typical 1H-NMR spectra of CL and PCL. | ||
Information about the chain end groups of synthesized macromolecules were obtained from matrix-assisted laser desorption and ionisation time of flight mass spectroscopy (MALDI-TOF MS – Bruker Daltonics Ultraflex II in positive reflectron mode) analysis. Relative populations of chains initiated by water or by β-CD were estimated by the following equation:
 | (3) |
 is the area of the signal related to the macromolecules initiated by water having Mi as molecular weight and
is the area of the signal related to the macromolecules initiated by water having Mi as molecular weight and  is the area of the signal related to the macromolecules initiated by β-CD having Mi as molecular weight. When cyclic chains were detected, also their population was considered in eqn (3) adding the corresponding term
is the area of the signal related to the macromolecules initiated by β-CD having Mi as molecular weight. When cyclic chains were detected, also their population was considered in eqn (3) adding the corresponding term  to the denominator.
to the denominator.
Solubility tests of selected samples were done in a Soxhlet extractor using water or THF, close to their boiling points, as solvents. Infrared spectra of samples recovered from Soxhlet extraction were recorded on a Perkin-Elmer Spectrum 2000 Explorer FTIR with an averaging of 30 scans at a resolution of 1 cm−1 using a near-IR fast recovery deuterated tryglicine sulphate detector. Analyses were performed on the polymer powder mixed with anhydrous KBr and then compressed to produce solid pellet.
3. Results and discussion
3.1 Ring opening polymerization of ε-caprolactone under pressure catalysed by wet β-cyclodextrin
A set of polymerization experiments was carried out at 393 K using CL dried on molecular sieves as model monomer both in the absence and in the presence of β-CD (Table 1).| Exp. | β-CD | [β-CD]/[CL] | Inert | P (MPa) | X (%) | DPn |
|---|---|---|---|---|---|---|
| a [β-CD]/[CL], molar ratio between cyclodextrin and monomer; P, reaction pressure; X, monomer conversion; DPn, number average degree of polymerization of synthesized PCL (1H-NMR determined), n.d.: not detectable. Reaction temperature 393 K, reaction time 24 h.b Experiment carried out adding to the liquid monomer an amount of water corresponding to 10% w/w with respect to the mass of the β-CD usually loaded. | ||||||
| 1 | — | 0 | N2 | 0.1 | n.d. | n.d. |
| 2 | — | 0 | CO2 | 12.0 | n.d. | n.d. |
| 3 | Dried | 1/101 | N2 | 0.1 | <1 | n.d. |
| 4 | Dried | 1/100 | CO2 | 11.5 | 5 | 3 |
| 5 | Wet | 1/100 | CO2 | 0.1 | 4 | n.d. |
| 6 | Wet | 1/102 | CO2 | 12.5 | 98 | 12 |
| 7 | Wet | 1/105 | N2 | 13.0 | 99 | 12 |
| 8 | Wet | 1/103 | Ar | 12.5 | 98 | 11 |
| 9 | Driedb | 1/101 | CO2 | 12.0 | 6 | 3 |
| 10 | —b | 0 | CO2 | 11.0 | 10 | 5 |
Differently from what performed by Harada and co-workers, who tested the cyclodextrin after drying under vacuum, we used both dried and native β-CD whose water content was estimated through thermogravimetric analyses to be comprised between 9 and 12% w/w.
At operative temperature and pressure adopted in this study both CL26 and PCL33 are negligible soluble in supercritical carbon dioxide. Moreover native β-CD must be peracetylated to exhibit appreciable solubility in scCO2.28 On these bases the polymerization occurs in the bulk monomer phase. This must be true also when permanent gases such as nitrogen and argon are used to pressurize the system.
When no cyclodextrin was loaded in the reactor, no monomer conversion was detected by 1H-NMR analyses both at room pressure and when the vessel was pressurized with CO2 at 12.0 MPa (Exp. 1 and 2, Table 1).
After addition of dried β-CD, that at room conditions was insoluble in the liquid monomer thus forming a solid suspension in the reactor, values of monomer conversion lower than 5% were estimated from 1H-NMR analyses of the liquid samples collected after 24 hours of reaction time both at room pressure and at 11.5 MPa (Exp. 3 and 4 Table 1).
These results are in agreement with those obtained by Harada et al. that reached PCL yields of 15% after 72 hours of polymerization at 100 °C and room pressure using a monomer to β-CD molar ratio of 5.16 Similar results were obtained using β-CD with its native water content at room pressure (Exp. 5 Table 1).
Differently, when the ROP was carried out using not dried β-CD in a reaction system pressurized at 12.5 MPa with CO2, using a CL/β-CD molar ratio as high as 102, a monomer conversion of 98% and a polyester having number average degree of polymerization of 12 were obtained after 24 hours of reaction time (Exp. 6 Table 1). It is known that scCO2 has a plasticizing effect on PCL23–25,34 that induces an enhancement of the chain mobility. To verify if the activation effect of the carbon dioxide pressure may be related to this effect we repeated the ROP of CL with the same recipe of Exp. 6 Table 1 but using N2 or Ar as pressurizing agent (Exp. 7 and 8 Table 1). In both cases monomer conversion and number average molecular weight were substantially the same than that obtained with scCO2 thus indicating that the pressure enhancement is the main factor affecting the performances of the ROP.
The nature of end groups of the synthesized polyester chains was investigated by MALDI-TOF.
From the spectra two populations of macromolecular chains were detected one having β-CD and –OH as terminal groups and the other having –COOH and –OH (Fig. 2a–d). The share of the two polymer families obtained in selected experiments are reported in Table 2.
 | ||
| Fig. 2 Maldi-TOF spectra of samples taken from the reactor at the end of selected polymerization runs reported in Table 1. (a) Exp. 4 Table 1; (b) Exp. 6 Table 1; (c) Exp. 7 Table 1; (d) Exp. 8 Table 1. | ||
In the case of the experiment carried out under pressure using dried CD, that led to monomer conversion of 5% after 24 hours, end capped β-CD oligomeric chains constituted 64% of the total product and 36% of PCL chains were initiated by water (Entry 1, Table 2).
When wet CD was used, water initiated chains were always the dominant population since their share was 70–78% under pressure of Ar or N2 and increased to 99% when scCO2 was used (Entries 2–4, Table 2).
PCL chains having β-CD as end group should be formed by the same polymerization mechanism proposed by Harada and co-workers that proposed the formation of an inclusion complex between the CD and the cyclic ester that activates the carbonyl group for nucleophilic attack of one hydroxyl group in position 2 of the CD.15–18 Moreover it was found that β-CD forms inclusion complexes with ε-CL in water solution and promotes its hydrolysis to the corresponding α,ω-hydroxyacid.35
This behaviour is similar to that of lipases that, in living systems, can catalyse the hydrolysis of esters in water but also the ring opening polymerization of cyclic esters in organic solvents or in the bulk monomer leading to formation of hydroxyl carboxyl terminated chains.36 For these enzymes, it is generally accepted that the hydroxyl group of serine 105 makes a nucleophilic attack on the carbonyl of the cyclic ester, leading to the formation of a linear acyl complex that is deacylated by a water molecule bound to the hydration shell of the enzyme thus leading to the formation of the α,ω-hydroxyacid. The latter hydroxyl bearing specie can deacylate another enzyme-activated monomer complex thus leading to the formation of dimeric species that can further act as deacylating agents thus propagating the chain.37
To obtain more information on the role of wet β-CD in the ROP of CL under CO2 pressure, we studied the effect of polymerization time on the number average degree of polymerization at long polymerization times (Table 3).
| Exp. | [β-CD]/[CL] | P (MPa) | Time (h) | X (%) | DPn | ωH2O | ωCD (%) | ωcyclic (%) |
|---|---|---|---|---|---|---|---|---|
| a [β-CD]/[CL], molar ratio between cyclodextrin and monomer; P, reaction pressure; X, monomer conversion; DPn, number average degree of polymerization of synthesized PCL (1H-NMR determined). ωH2O, ωCD and ωcyclic are the fractions of chains initiated by water, CD and cyclic respectively. Reaction temperature 393 K. | ||||||||
| 1 | 1/101 | 12.0 | 6 | 20 | 2 | 96 | 4 | n.d. |
| 2 | 1/100 | 12.3 | 24 | 98 | 12 | 99 | 1 | n.d. |
| 3 | 1/96 | 12.9 | 48 | 99 | 23 | 85 | 10 | 5 |
| 4 | 1/98 | 12.5 | 72 | 99 | 27 | 84 | 9 | 7 |
We found a trend of monomer conversion and DPn that is typical of a step growth polymerization as, even if quantitative monomer conversion was reached after 24 h, longer reaction time made possible to increase the average degree of polymerization from 12 to 27 (Exp. 2–4, Table 3).
Polymer obtained at different polymerization times were analysed by MALDI-TOF. In all experiments, the main population of chains were hydroxyacid terminated. The fraction of these polyester chains decreased from values higher than 95% to 84–85% when polymerization times higher than 24 h were used. This decrement was accompanied by an increase in the population of CD terminated chains (Fig. 3b). Since monomer was almost consumed at these high reaction times aforementioned chains must be formed by transesterification reactions. Quite interestingly also macrocyclic chains with number of repeat units changing from 6 to 13 were observed in the MALDI spectra (Fig. 3c). The formation of such macrocycles is another element of similarity with lipase catalyzed ROP of CL and it could be due to a back-biting reaction in which the terminal hydroxyl of a CD bearing linear oligomers acts as deacylating agent of the polyester acyl complex.
 | ||
| Fig. 3 Plot of percentage areas of the MALDI-TOF signals as a function of number average degree of polymerization for water terminated (a), CD terminated (b) and cyclic (c) chains obtained in experiments reported in Table 3. | ||
All reported experimental results indicate that in the presence of wet β-CD a reaction mechanism similar to that proposed for lipase catalyzed ROP of cyclic esters is operative. Indeed it seems that hydroxyl group in position 2 of the CD can mimic the role of the hydroxyl group of serine 105. Both OH– groups bring a nucleophilic attack on the carbonyl of the cyclic ester leading to the formation of a linear acyl complex, while inner basket water resemble the role of water molecules bound to the hydration shell of the enzyme as deacylating agent thus leading to the formation of an α,ω-hydroxyacid. Longer chain formation can be explained considering that hydroxyl groups of linear α,ω-hydroxyacids can act themselves as deacylating agents.
To find other experimental data supporting this hypothesis we performed a set of experiments to study the interaction of wet β-CD with commercial PCL chains whose number average degree of polymerization was estimated to be 44 by 1H-NMR spectroscopy in CDCl3 (Table 4). If the analogy with enzyme should be valid, the hydroxyl group of CD should also catalyse the hydrolysis of preformed polyester chains and CD terminated chains should be detected in the MALDI spectra. When commercial PCL was contacted with scCO2 at 13 MPa for 24 h no modification of 1H-NMR determined DPn was found. Differently when the same experiment was repeated in the presence of wet-CD, added with a molar ratio 1/96 with respect to the amount of polymer repeat units, DPn decreased to 26 thus indicating that the hydrated CD can catalyse the hydrolysis of preformed PCL chains (Table 4). Quite interestingly, when the hydrolysed polymer was analysed by MALDI-TOF a population of CD terminated chains was detected thus supporting aforementioned similarity.
| [β-CD]/[CL] | Inert | P (MPa) | DPn |
|---|---|---|---|
| a [β-CD]/[CL], molar ratio between cyclodextrin and monomer; P, reaction pressure; DPn, number average degree of polymerization of PCL (1H-NMR determined). Reaction temperature 393 K, reaction time 24 h.b Number average degree of polymerization of commercial PCL. | |||
| 0 | — | — | 44b |
| 0 | CO2 | 13.0 | 44 |
| 1/96 | CO2 | 13.0 | 26 |
The experimental results obtained studying the effect of polymerization time and the behaviour of PCL chains contacted with β-CD under pressure are all compatible with the possibility that the wet oligosaccharides can work as a chemzyme in the ROP of CL.38
It is interesting to underline that when the ROP was performed with dried β-CD no significant variation of the value of CL conversion was observed working at room pressure or in pressurized reactors (Exp. 3 and 4 Table 1). These results indicate that, beside pressure, another essential element in determining the enhancement of the polymerization rate is water and to study its effect in the CD activated polymerization, we performed a control experiment with dried β-CD at the same operative conditions of Exp. 6 Table 1 but adding to the monomer an amount of water corresponding to 10% w/w with respect to the mass of CD loaded in the reactor (Exp. 9 Table 1). In these conditions the outcome of the polymerization was substantially the same than that obtained with dried β-CD. One additional test was carried out using the same recipe but in the absence of any CD. In this case monomer conversion reached 10% after 24 hours of reaction time (Exp. 10 Table 1).
Quite interestingly, a behaviour similar to that found in this study has been recently reported for the heterodomino reaction between 2-phenylbut-3-yn-2-ol and p-cresol in water at 60 °C.39 Hydrated β-CD was found to be an effective catalyst for this reaction while, dry β-CD was not active at all in spite of the fact that both compounds were dissolved in an aqueous reaction medium at reaction conditions. It is important to mention that when β-CD hydrate was dried at 130 °C for 4 h, conditions at which the loss of water was complete, and added to the aqueous reaction medium, the aforementioned reaction did not occur at all.39 This behavior was attributed to a key role of water molecules inside the CD basket in the activation of the reaction. Indeed we can consider that native β-CD rapidly equilibrates with atmospheric humidity at room temperature and it is organized in a crystalline structure that at normal ambient conditions (18 °C and about 50% humidity) has the composition β-CD·10.5H2O with about 5 water molecules between the β-CD units and about 5.5 inside the β-CD cavity.40,41 Calorimetric studies have shown that the latter water molecules have an energetic content much higher than that of bulk water,42,43 moreover it has been shown that β-CD can form inclusion complexes with ε-CL both in aqueous phase35 and in the bulk monomer.16 In the case of formation of inclusion complexes in water solution, the release of high energy water molecules from the β-CD cavity into the bulk of the aqueous phase, also termed hydrophobic effect, is considered as a significant contribution to the complexation thermodynamics.9 Much less informations are available on the driving force for inclusion complex in solid state or organic solvents.
On the other hand, the energetic gain in the transfer of water molecules from the basket to the bulk of the monomer phase should be significantly lower than that in aqueous systems and it seems reasonable to postulate that the hydrophobic effect should play a minor role in determining the driving force for complexation in liquid CL. For these reasons when the cyclic ester is complexed by the CD, residual water molecules of high energetic content could be still present inside the basket playing a role in initiating the ROP.
In this intellectual frame, the experimental evidence that dried β-CD is not efficient as polymerization catalyst even if water is added to the monomer phase, could be explained by a not favourable partitioning of water between the liquid monomer phase and the basket of the CD owing to the hydrophobic nature of the cavity and the large excess of CL molecules.
From the experimental results reported in Table 1 it can be anyway observed that wet β-CD used at room pressure led to monomer conversion of just 4% after 24 hours and only working at 12 MPa we reached almost quantitative conversion of the cyclic ester (Exp. 5 and 6). These results indicate that the significant enhancement of the polymerization kinetics must be attributed, beside to the presence of water inside the basket of CD, also to the effect of pressure.
It has been demonstrated that the operative pressure can affect inclusion behaviour of native cyclodextrins in aqueous solutions.44,45 In particular when the sign of partial molar volume variation upon complexation ΔV is negative, higher pressure can induce an increase of the equilibrium constant according to eqn (4) where K is the inclusion equilibrium constant, R the gas constant and T and P the operative temperature and pressure respectively.
 | (4) |
In the polymerization system here considered, that is initially characterized by a suspension of solid hydrated β-CD in the liquid monomer, only dissolved β-CD or surface located units in crystallite should be candidate to activate the polymerization since cavity of hydrated units incorporated in crystallites should be inaccessible to the monomer owing to their fishbone type packing.41
The positive effect of higher pressure could be tentatively related to a contraction of partial molar volume upon complexation of CL by β-CD that induces an increase of the value of the inclusion constant K that enhances the apparent solubility of the β-CD in the molten polymerization medium thus bringing to an augmentation of the effective concentration of the cyclic oligosaccharide.
3.2 Formation of polyrotaxane between PCL chains and wet β-cyclodextrin
Since, according aforementioned considerations, only dissolved free CD units should be considered as active catalysts it seems relevant to underline that β-CD can form inclusion complexes also with the polyester chains46 so that the effective local concentration of the cyclic oligosaccharide during the polymerization can be affected by this additional equilibrium.When PCL samples synthesised in the presence of wet β-CD were mixed with THF we always found an insoluble portion. Free CD is well known to be insoluble in aforementioned solvent and then it must be certainly a component of this fraction. On the other hand it is possible that also PCL chains complexed by the CD are insoluble in the organic solvent because, in-spite of the absence of terminal stoppers that prevent the decomposition of the supramolecular aggregate,47 it remains stable in the time window necessary for sample preparation owing to a too slow kinetics of diffusion of the CD out of the polymer backbone. To study this possibility we performed FTIR analysis of the insoluble fraction obtained by filtration. In Fig. 4 the FTIR spectra of free β-CD (spectrum a) and of a polymer sample synthesised in the presence of β-CD, divided in portion insoluble (spectrum b) and soluble (spectrum c) in THF are shown. The soluble portion was characterised by a peak at 1730 cm−1, which can be attributed to PCL.48 It is interesting to note that the insoluble portion was not constituted by free β-CD only: there was, in fact, the peak of the carbonyl at 1730 cm−1 and a not very pronounced peak at 1644 cm−1 that is present also in spectrum of β-CD. The presence of the peak due to the carbonyl can be considered an indication that part of PCL chains are made insoluble in THF by complexation with β-CD.
 | ||
| Fig. 4 FTIR spectra of β-CD and of samples obtained from the final product of ROP carried out under CO2 pressure in the presence of β-CD. Pure β-CD (a), sample taken from the portion insoluble in THF (b), sample taken from the portion soluble in THF (c). | ||
To have additional information on the structure of the synthesised polymer, we analysed two different residual samples obtained after Soxhlet extractions with boiling THF or water respectively (Fig. 5): the first is a good solvent for PCL, the second for β-CD. The residue obtained after the extraction with THF (spectrum b) presented the carbonyl peak at 1730 cm−1 and a little peak at 1644 cm−1. The insoluble fraction obtained after the extraction with water, instead, was characterised only by the carbonyl peak at 1730 cm−1, showing that a good solvent of the CD is necessary to remove completely it from PCL chains.
 | ||
| Fig. 5 FTIR spectra of β-CD and of samples obtained from the final product of ROP carried out under CO2 pressure in the presence of β-CD after Soxhlet extraction with THF or water. Pure β-CD (a), sample taken from the fraction insoluble in THF (b), sample taken from the fraction insoluble in water (c). | ||
3.3 β-CD catalysed ROP of ε-CL activated by pressure: effect of the catalyst concentration and of the polymerization temperature
Some polymerizations were addressed to investigate the effect of the concentration of wet β-CD and of reaction temperature on the rate of the ROP and the average molecular weight of polymer chains synthesized in pressurized systems (Table 5).| Exp. | [β-CD]/[CL] | Inert | T (K) | Time (h) | X (%) | DPn | Np × 103 (mmol mmol−1) |
|---|---|---|---|---|---|---|---|
| a [β-CD]/[CL], molar ratio between cyclodextrin and monomer; T, polymerization temperature; X, monomer conversion; DPn, number average degree of polymerization (1H-NMR determined). Operative pressure = 12.0–12.5 MPa. | |||||||
| 1 | 1/21 | CO2 | 393 | 16 | 98 | 4 | 245 |
| 2 | 1/104 | CO2 | 393 | 16 | 43 | 8 | 54 |
| 3 | 1/101 | Ar | 393 | 4 | 6 | 2 | 30 |
| 4 | 1/100 | Ar | 408 | 4 | 39 | 5 | 78 |
| 5 | 1/101 | Ar | 423 | 4 | 93 | 14 | 66 |
To study the effect of the concentration of CD we performed experiments at two different CD/CL molar ratios, namely 1/104 and 1/21, by keeping fixed polymerization temperature and pressure at 393 K and 12 MPa respectively. When the lower CD concentration was used a monomer conversion of 43% was obtained after 16 hours and average number of repeat units incorporated in PCL chains was 8 (Exp. 2, Table 5). By increasing fivefold the CD concentration the monomer conversion reached a value of 98% and the average length of synthesized chains was estimated to be one half of that of chains obtained when CD/CL was 1/104 (Exp. 1, Table 5).
The faster polymerization rate and the lower average molecular weight of synthesized polyesters are indicative that a higher concentration of active growing chains was generated when the concentration of β-CD was increased. If we estimate from 1H-NMR data the number of polymer chains per mmol of monomer (Np) it is possible to observe that this number decreases by a factor five when the value of CD/CL is changed from 1/21 to 1/104. This result is a strong indication that the number of chains is correlated with the CD concentration thus supporting the hypothesis that under adopted conditions it behaves more as a catalyst rather than an initiator.
The effect of temperature was studied performing polymerization experiments at 393, 408 and 423 K. In all experiments, the reactor was pressurized with Ar at 12.0–12.5 MPa and molar ratio CD/CL close to 1/100 was adopted. Higher polymerization temperature led to higher monomer conversion and number average polymerization degree.
Also in these experiments Np was estimated from 1H-NMR data. We found that Np increased significantly when the temperature changed from 120 to 135 °C while remained substantially unchanged when the temperature was further augmented to 150 °C. As previously reported, the role of β-CD strongly resembles that of a lipase enzyme since it can cumulate molecular recognition, related to the complexation reaction with the cyclic ester, and specific reactivity arising from the nucleophilic secondary hydroxyl functional groups that attack the included molecule opening its ring. These steps can be considered the initiation steps of the polyester chain while deacylation by water or by hydroxyl bearing monomers and/or oligomers are responsible for chain growth.37 In this scenario the trends of Np and of the average degree of polymerization with temperature are coherent with the hypothesis that the rate of the chain growth steps increases with temperature more rapidly than the rate of the initiation steps.
4. Conclusions
Ring opening polymerization of ε-caprolactone was carried out in the presence of both dried and wet β-CD under pressure of CO2, N2 or Ar. After 24 hours of polymerization time at 393 K, dried β-CD gave polymer yields lower than 5% at all investigated operative conditions even when water was added to the liquid monomer prior to the polymerization experiment. Similar results were obtained when wet β-CD was used at room pressure. Differently when the polymerization was carried out at 11–13 MPa, with a molar ratio CD/CL close to 1/100, a significant acceleration of the polymerization rate was observed since quantitative monomer conversion was reached after 24 h at 393 K with all adopted gases. Experiments carried out at longer polymerization time have shown that molecular weight of synthesized polymers increases even in the absence of cyclic monomer. Moreover wet CD is also able to catalyse the hydrolysis of preformed PCL chains and CD terminated chains are generated in this process. Both cyclodextrin and water end-capped polyester chains were detected, the former having the highest share in the distribution. Collected results indicate that when pressure is enhanced, wet CD can catalyse the ROP polymerization of ε-CL with a mechanism that has strong similarity with that of lipase catalyzed ROP of cyclic esters and that inner basket water must play a role in the initiation of the polymerization probably during the formation of the inclusion complex between the monomer and the cyclic oligosaccharide.Acknowledgements
The financial support of University of Palermo is gratefully acknowledged. P. L. is Research Associate by the FRS-FNRS. B. G. thanks the “Région Wallonne” for its financial support in the frame of the SINOPLISS project (FEDER program 2007–2013). P. L. and B. G. thank the Belgian Science Policy for financial support in the frame of the Interuniversity Attraction Poles Program (P7/05)-Functional Supramolecular Systems (FS2). Authors are grateful to one of the reviewers for the constructive comments.References
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841 CrossRef CAS.
- A. C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466 CrossRef CAS PubMed.
- C. Jérôme and P. Lecomte, Adv. Drug Delivery Rev., 2008, 60, 1056 CrossRef PubMed.
- M. S. Lindblad, Y. Liu, A. C. Albertsson, E. Ranucci and S. Karlsson, Adv. Polym. Sci., 2002, 157, 139 CrossRef CAS.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093 CrossRef CAS.
- R. A. Gross, A. Kumar and B. Kalra, Chem. Rev., 2001, 101, 2097 CrossRef CAS PubMed.
- S. Matsumara, Adv. Polym. Sci., 2006, 194, 95–132 CrossRef.
- Y. Yang, Y. Yu, Y. Zhang, C. Liu, W. Shi and Q. Li, Process Biochem., 2011, 46, 1900 CrossRef CAS.
- M. V. Rekharsky and Y. Inoue, Chem. Rev., 1998, 98, 1875 CrossRef CAS PubMed.
- I. Tabushi, Acc. Chem. Res., 1982, 15, 66 CrossRef CAS.
- W. B. Motherwell, M. J. Bingham and Y. Six, Tetrahedron, 2001, 57, 4663 CrossRef CAS.
- J. Bjerre, C. Rousseau, L. Marinescu and M. Bols, Appl. Microbiol. Biotechnol., 2008, 81, 1–11 CrossRef CAS PubMed.
- Y. Miao and P. Zinck, Polym. Chem., 2012, 3, 1119 RSC.
- Y. Miao, C. Rousseau, A. Mortreux, P. Martin and P. Zinck, Polymer, 2011, 52, 5018 CrossRef CAS.
- Y. Takashima, M. Osaki and A. Harada, J. Am. Chem. Soc., 2004, 126, 13588 CrossRef CAS PubMed.
- M. Osaki, Y. Takashima, H. Yamaguchi and A. Harada, Macromolecules, 2007, 40, 3154 CrossRef CAS.
- M. Osaki, Y. Takashima, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2007, 129, 14452 CrossRef CAS PubMed.
- A. Harada, M. Osaki, Y. Takashima and H. Yamaguchi, Acc. Chem. Res., 2008, 41, 1143 CrossRef CAS PubMed.
- M. Osaki, Y. Takashima, H. Yamaguchi and A. Harada, Org. Biomol. Chem., 2009, 7, 1646 CAS.
- M. Osaki, Y. Takashima, H. Yamaguchi and A. Harada, J. Org. Chem., 2009, 74, 1858 CrossRef CAS PubMed.
- A. Harada, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4469 CrossRef CAS.
- Y. Takashima, M. Osaki, Y. Ishimaru, H. Yamaguchi and A. Harada, Angew. Chem., Int. Ed., 2011, 50, 7524 CrossRef CAS PubMed.
- G. A. Leeke, J. Cai and M. Jenkins, J. Chem. Eng. Data, 2006, 51, 1877 CrossRef CAS.
- Z. Lian, S. A. Epstein, C. W. Blenk and A. D. Shine, J. Supercrit. Fluids, 2006, 39, 107 CrossRef CAS.
- C. A. Kelly, S. H. Murphy, G. A. Leeke, S. M. Howdle, K. M. Shakesheff and M. J. Jenkins, Eur. Polym. J., 2013, 49, 464 CrossRef CAS.
- F. Stassin and R. Jérome, Chem. Commun., 2003, 232 RSC.
- R. Span and W. Wagner, J. Phys. Chem. Ref. Data, 1996, 25, 1509 CrossRef CAS.
- V. K. Potluri, A. D. Hamilton, C. F. Karanikas, S. E. Bane, J. Xu, E. J. Beckman and R. M. Enick, Fluid Phase Equilib., 2003, 211, 211 CrossRef CAS.
- D. Bradley, G. Williams and M. Lawton, J. Org. Chem., 2010, 75, 8351 CrossRef PubMed.
- R. T. MacDonald, S. K. Pulapura, Y. Y. Svirkin, R. A. Gross, D. L. Kaplan, J. Akkara, G. Swift and S. Wolk, Macromolecules, 1995, 28, 73 CrossRef CAS.
- A. Kiersnowski, P. Dabrowski, H. Budde, J. Kressler and J. Piglowski, Eur. Polym. J., 2004, 40, 2591 CrossRef CAS.
- A. Córdova, T. Iversen, K. Hult and M. Martinelle, Polymer, 1998, 39, 6519 CrossRef.
- S. R. Rojo, A. Martín, E. Sáez Calvo and M. J. Cocero, J. Chem. Eng. Data, 2009, 54, 962 CrossRef CAS.
- M. A. Fanovich and P. Jaeger, Mater. Sci. Eng., Proc. Conf., 2012, 32, 961 CrossRef CAS.
- Y. Takashima, Y. Kawaguchi, S. Nakagawa and A. Harada, Chem. Lett., 2003, 32, 1122 CrossRef CAS.
- S. Kobayashi, Macromol. Rapid Commun., 2009, 30, 237 CrossRef CAS PubMed.
- A. Córdova, T. Iversen, K. Hult and M. Martinelle, Polymer, 1998, 39, 6519 CrossRef.
- J. Bjerre, C. Rousseau, L. Marinescu and M. Bols, Appl. Microbiol. Biotechnol., 2008, 81, 1 CrossRef CAS PubMed.
- A. Ghatak, S. Khan and S. Bhar, Adv. Synth. Catal., 2016, 358, 435 CrossRef CAS.
- T. Steiner, G. Koellner, S. Ali, D. Zakim and W. Saenger, Biochem. Biophys. Res. Commun., 1992, 188, 1060 CrossRef CAS PubMed.
- T. Steiner and G. Koellner, J. Am. Chem. Soc., 1994, 116, 5122 CrossRef CAS.
- I. V. Terekhova, R. De Lisi, G. Lazzara, S. Milioto and N. Muratore, J. Therm. Anal. Calorim., 2008, 92, 285 CrossRef CAS.
- G. Wenz, J. Org. Chem., 2012, 8, 1890 CAS.
- S. M. Hoenigman and C. E. Evans, Anal. Chem., 1997, 69, 2136 CrossRef CAS PubMed.
- Y. Sueishi, H. Tobisako and Y. Kotake, J. Phys. Chem. B, 2004, 108, 12623 CrossRef CAS.
- Y. Kawaguchi, T. Nishiyama, M. Okada, M. Kamachi and A. Harada, Macromolecules, 2000, 33, 4472 CrossRef CAS.
- A. Harada, A. Hashidzume, H. Yamaguchi and Y. Takashima, Chem. Rev., 2009, 109, 5974 CrossRef CAS PubMed.
- T. Elzein, M. Nasser-Eddine, C. Delaite, S. Bistac and P. Dumas, J. Colloid Interface Sci., 2004, 273, 381 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |