
The interaction of NOTA as a bifunctional chelator with competitive alkali metal ions: a DFT study†
Abstract
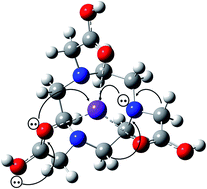
1,4,7-Triazacyclononane-1,4,7-triacetic acid (NOTA) is a key chelator for radiolabelling pharmaceuticals. The ability of alkali metals in the human body to complex with NOTA and compete with radiometals can influence the radiolabelling process. The focus of the present work is to evaluate the NOTA–alkali metal complexation with density functional theory (B3LYP functional) using the 6-311+G(2d,2p) basis set for Li+, Na+ and K+ and Def2-TZVPD for Rb+. Two NOTA–ion conformations are reported in the study: ‘A’ where six NOTA hetero atoms (N, O) are in close proximity to the cation, and ‘B’, where four NOTA hetero-atoms interact with the cation. Interaction and relaxation energies, Gibbs free energies and entropies show that the stability of NOTA–ion complexes decreases down the group of the periodic table. Implicit water solvation affects the NOTA–ion complexation, causing a decrease in the stability of the system. NBO analysis performed through the natural atomic charges (NAC) and second order perturbation analysis reveals charge transfer between NOTA and alkali metals. The theoretical 1H NMR chemical shifts of NOTA, in vacuum and water media, are in good agreement with experiments, these values being influenced by the presence of the ions, which have a deshielding effect on the protons of NOTA. Global scalar properties, such as HOMO/LUMO energies, ΔELUMO–HOMO gap, and chemical hardness and softness, show that the chemical stability of NOTA–alkali metal complexes decreases down the periodic table. This study sheds light on the impact of competing alkali metal ions to the radiolabelling efficiency of NOTA.
 Please wait while we load your content...
Please wait while we load your content...

