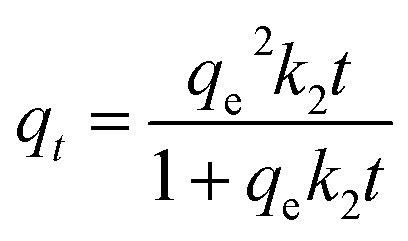DOI:
10.1039/C6RA20188A
(Paper)
RSC Adv., 2016,
6, 101198-101207
Facile one-pot synthesis of carbon incorporated three-dimensional hierarchical TiO2 nanostructure for highly efficient pollutant removal†
Received
10th August 2016
, Accepted 10th October 2016
First published on 10th October 2016
Abstract
Herein, a carbon incorporated three-dimensional (3D) hierarchical TiO2 nanostructure (CRF-TiO2) was successfully synthesized via a facile one-pot route by coupling TiCl3 hydrolysis–oxidation with resorcinol–formaldehyde (RF) reaction without any surfactant. The fabricated CRF-TiO2 showed an interesting 3D hierarchical and mesoporous structure, possessed relatively large specific surface area and excellent light absorption capability, and the TiO2 in the final product was an anatase–rutile mixture with mature crystallinity. All of the above characteristics endow the prepared CRF-TiO2 with superior adsorption capability and photocatalytic activity compared to Degussa P25. As a result, with the presence of CRF-TiO2, pollutants in water could be rapidly separated from the aqueous phase via adsorption, which can ideally meet the requirements of fast water purification, and then the pollutants would be efficiently decomposed by a photocatalysis process under both UV and visible light irradiation to completely fulfill the pollutant removal target. The synthetic strategy for CRF-TiO2 employed in this study provides an effective solution for the synthesis of carbon incorporated 3D hierarchical TiO2 architectures, and can introduce new insights to design TiO2-based photocatalysts for practical environmental purification applications.
Introduction
Facing the serious global challenge of scarcity of clean water and increasingly stringent water standards, photocatalysis, which can decompose almost all organic pollutants in water under mild conditions without producing secondary pollution, has attracted considerable research attention around the world in recent years.1,2 Among the various photocatalysts investigated, TiO2 has been proved to be one of the most promising photocatalysts owing to its low cost, wide availability, non-toxicity, good stability and environmentally friendly nature.3 However, the wide application of TiO2 in practical water purification is seriously hampered by (1) the low pollutant degradation rate caused by the rapid recombination of photoexcited electron–hole pairs in TiO2 and limited contact of TiO2 with pollutants,4,5 (2) poor efficiency under solar light irradiation due to the wide band gap of TiO2,6,7 and (3) difficult photocatalyst recovery after use since most of the TiO2 under investigation is at the nanoscale for the sake of high photocatalytic activity.8
Recently, micron-sized 3D hierarchical TiO2 nanostructures composed of one-dimensional (1D) TiO2, such as nanorods or nanowires, have received much attention and been widely applied in dye sensitized solar cells (DSSCs), lithium-ion batteries (LIBs) and photocatalysis.9–12 Lin et al. successfully prepared a well-defined 3D hierarchical rutile TiO2 architecture for DSSCs, and a remarkable overall conversion efficiency of 8.6% was recorded.9 Wang et al. synthesized a chrysanthemum-like TiO2 nanostructure with controllable morphology, and a superior capacity up to 309.3 mA h g−1 was achieved in LIBs test.10 The outstanding performance of 3D hierarchical TiO2 based applications can be ascribed to the efficient charge transfer facilitated by 1D TiO2 building blocks, enlarged surface area and enhanced light harvesting capability of 3D hierarchical TiO2,9–11 all of which also result in excellent performance of 3D hierarchical TiO2 in pollutant photodegradation, where Bai et al. reported that the degradation rate of acid orange 7 and phenol by a 3D hierarchical TiO2 with large percentage of (110) facet exposure was much faster than that of Degussa P25.13 In addition, the dimensions of 3D hierarchical TiO2 nanostructures are on the microscale, which makes it possible to recover them after use by gravity sedimentation or simple membrane filtration.11 Therefore, from the viewpoints of high activity and easy recovery, 3D hierarchical TiO2 gives new insights for application of photocatalysis in practical water purification.
Using carbonaceous materials to enhance the photocatalytic activity and visible light response of TiO2 has attracted considerable attention due to the unique and controllable structural and electrical properties of the products.5 Activated carbon–TiO2 composites are reported to show a synergistically enhanced photocatalytic activity over TiO2 alone, which is attributed to the efficient adsorption of pollutants on activated carbon followed by mass transfer to TiO2 through their common interfaces,14 which provide more chances and extend the reaction time for the pollutants and their intermediates to contact with TiO2. For carbon nanotubes–TiO2 composites, researchers found that carbon species may act as photosensitizers, transferring electrons to the TiO2 and extending TiO2 photocatalytic activity into the visible light range.15 Carbonaceous materials do contribute to improving the pollutant removal capability and extending the light absorption range of TiO2, however, to date, most carbonaceous TiO2 has focused on TiO2 nanoparticles,14,16,17 and only a few carbon based hierarchical TiO2 have been reported.18,19 Moreover, in the limited existing reports, carbon was present in small amounts and doped in the lattice of hierarchical TiO2 crystal;18,19 as a result, on one hand, no obvious improvement of the adsorption capability of TiO2 could be observed, and on the other hand, the multistep synthesis process was too elaborate and complicated for large-scale production.
Hydrolysis and oxidation of TiCl3 has been reported to be able to produce urchin-like TiO2 submicron spheres via a simple synthesis route under mild reaction conditions, and the produced TiO2 exhibited excellent performance in LIBs.20 Meanwhile, the resorcinol–formaldehyde reaction is widely used in both research and the chemical industry to produce carbon aerogel microspheres with large specific surface areas and pore volumes. Interestingly, TiCl3 solution can catalyze the RF reaction; moreover, reaction conditions for both the TiCl3 conversion to TiO2 and the RF reaction show great compatibility,20,21 which signifies the great possibility of combining the two reactions. In this study, we try to proceed these two reactions synchronously, namely a facile one-pot RF + TiCl3 route, to synthesize a carbon incorporated 3D hierarchical TiO2 (CRF-TiO2) for photocatalytic pollutant removal from water. Reaction conditions of the RF + TiCl3 route were not harsh and operations were also normal, indicating that the RF + TiCl3 route is suitable for mass production.
A great amount of azo dyes wastewater from the textiles, printing and leather industries is discharged into the aquatic environment each year, posing great ecological and health threats to aquatic organisms and human beings.22 However, conventional water treatment processes, such as coagulation/flocculation or biological treatment, cannot ideally handle the dye wastewater due to the generation of secondary pollution and the non-biodegradable characteristics of azo dye.23 In recent years, TiO2 photocatalytic oxidation for dye wastewater treatment has attracted much attention owing to its great oxidation capability even for refractory organics.24 Herein, we chose the widely used azo dye, namely acid brilliant scarlet (GR), as the probe to evaluate the adsorption capability and photocatalytic activity of the synthesized CRF-TiO2, where the behavior and mechanism of GR adsorption on the CRF-TiO2 surface and GR photodegradation catalyzed by CRF-TiO2 under both UV and visible light irradiation were investigated in detail with Degussa P25 as a comparison.
Experimental
Materials
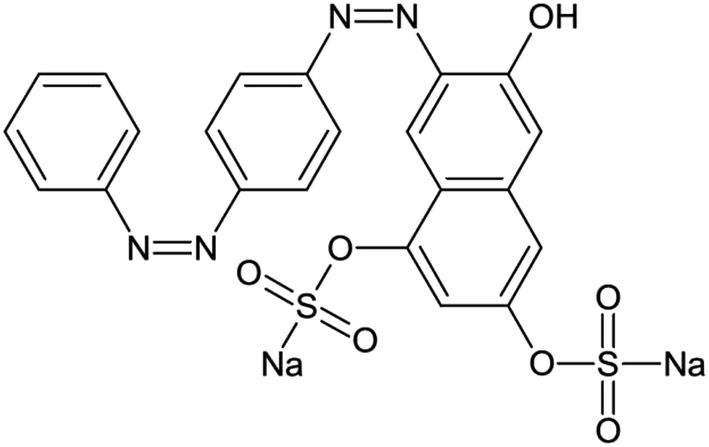
Resorcinol, formaldehyde (35 wt%) and TiCl3 (20 wt% TiCl3 dissolved in 3 wt% HCl solution) were all of analytical grade and purchased from Sinopharm Chemical Reagent Co., Ltd, China. All the chemicals were used without further purification. The molecular structure of GR (C22H14N4Na2O7S2, 556.49 g mol−1) is shown in Fig. 1. Deionized water was produced by a Milli-Q system and used systematically in this study.
 |
| | Fig. 1 Molecular structure of GR. | |
Synthesis
In a typical RF + TiCl3 synthetic procedure, 2.75 g of resorcinol and 4 mL of formaldehyde were first dissolved in 95 mL of deionized water successively, and 12 mL of TiCl3 was then added dropwise into the above solution under vigorous stirring. After 2 hours of polymerization reaction at 70 °C under continuous stirring, Ti/RF spheres were produced. The obtained precipitate was first washed with deionized water thoroughly, and then dried in an oven at 105 °C overnight; the obtained sample is denoted as RF–TiO2. The RF–TiO2 sample was further calcined at 500 °C for 2 hours with limited exposure to oxygen in a tube furnace, under a 20 vol% oxygen-containing argon atmosphere with a gas flow rate of 100 mL min−1, to improve the crystallinity of TiO2 and adjust the TiO2 content in the sample, and the finally synthesized carbon incorporated TiO2 sample is denoted as CRF-TiO2.
Characterization
The X-ray diffraction (XRD) pattern of the samples was obtained using a Bruker D8 Advance X-ray diffractometer with monochromated high-intensity Cu Kα radiation (λ = 1.5418 Å). Sample morphology was observed by field emission scanning electron microscopy (FESEM, JEOL JSM-6700F) and a high-resolution transmission electron microscope (HRTEM, JEOL JEM-2100F). Elemental mapping was detected by an energy dispersive X-ray spectrometer (EDS, Horiba EX-450) attached to a FESEM (Hitachi 4800). The thermo-gravimetric analysis (TGA) of the fabricated samples was conducted under atmospheric conditions using an SDT Q600 (TA Instruments, USA) from room temperature to 800 °C. X-ray photoelectron spectroscopy (XPS) analysis was carried out using a Thermo ESCALAB 250Xi equipped with standard monochromatic Al Kα excitation source (hν = 1486.6 eV), and all binding energies were referenced to C 1s at 284.8 eV. Diffuse-reflectance spectra of samples were recorded by UV-2550 UV-visible spectrophotometer (Shimadzu). The BET surface area and pore size distribution were determined using N2 adsorption–desorption isotherms at 77 K by an automated pore size and surface area analyzer (JW-BK122W, Beijing JWGB).
Adsorption tests
In a typical adsorption experiment, the photocatalyst (CRF-TiO2 or P25, 0.5 g L−1) was added into 100 mL of aqueous solution of GR in Erlenmeyer flasks with 200 rpm stirring at room temperature. The flasks were wrapped and covered by aluminum foil to minimize evaporation and eliminate dye degradation caused by light irradiation. In the kinetic study, the initial concentrations of GR were 10, 30 and 50 mg L−1. Samples were collected at stated time intervals and subsequently filtered through a Millipore filter (0.45 μm). The concentration of GR in the filtrate was determined by using a UV-2450 UV-visible spectrophotometer (Shimadzu) at its maximum absorption wavelength of 511 nm. Adsorption isotherm tests were performed using a series of dye solutions with different initial concentrations ranging from 5 to 80 mg L−1. Sampling was carried out after 24 h of mixing to ensure the equilibrium was approached, and the equilibrium adsorption capacity, qe (mg g−1), was calculated as follows:where C0 and Ce (mg L−1) are the initial and equilibrium concentration of dye, respectively, V (L) is the volume of dye solution and M (g) is the mass of adsorbent.
Photocatalytic tests
For the photocatalytic degradation of GR under UV irradiation, the apparatus employed consisted of a cylindrical Pyrex reactor exposed to air and an immersed UV lamp (14 W, 254 nm, Shanghai JG Special Lighting) as the light source. A fixed mass of photocatalyst (CRF-TiO2 or P25, dosage at 0.2 g L−1) was added into GR solution (C0 = 50 mg L−1) and stirred continuously. Before irradiation, the suspension was continuously stirred in the dark for 1 hour to allow a large proportion of adsorption of GR (about 80%) on the photocatalyst. The adsorption time here was determined based on both the adsorption kinetics study carried out above and practical application feasibility. After the UV light was switched on, sampling was carried out at given time intervals, and the residual GR concentration was determined at its maximum absorption wavelength of 511 nm, since no new bands appeared in the UV-visible absorbance spectra during the degradation. The residual total organic carbon (TOC) of the dye solution was also analyzed by total organic carbon analyzer (TOC-V CPH, Shimadzu) to determine the mineralization degree of the dye during photocatalytic degradation. For the photocatalytic tests under visible light irradiation, a short-arc xenon lamp (CHF-XM-500W, Beijing CT) equipped with a 420 nm cutoff filter was used as the light source. As a control, photolysis of GR without the addition of photocatalyst was also conducted under the same conditions.
It is worth noting that, in order to perform the photocatalytic kinetics study of CRF-TiO2 and P25 more precisely, the photocatalyst dosage employed in this study was set at 0.2 g L−1, which was much lower than some other reports (1 g L−1),25 to extend the photocatalytic reaction period.
Results and discussion
Characterizations of CRF-TiO2
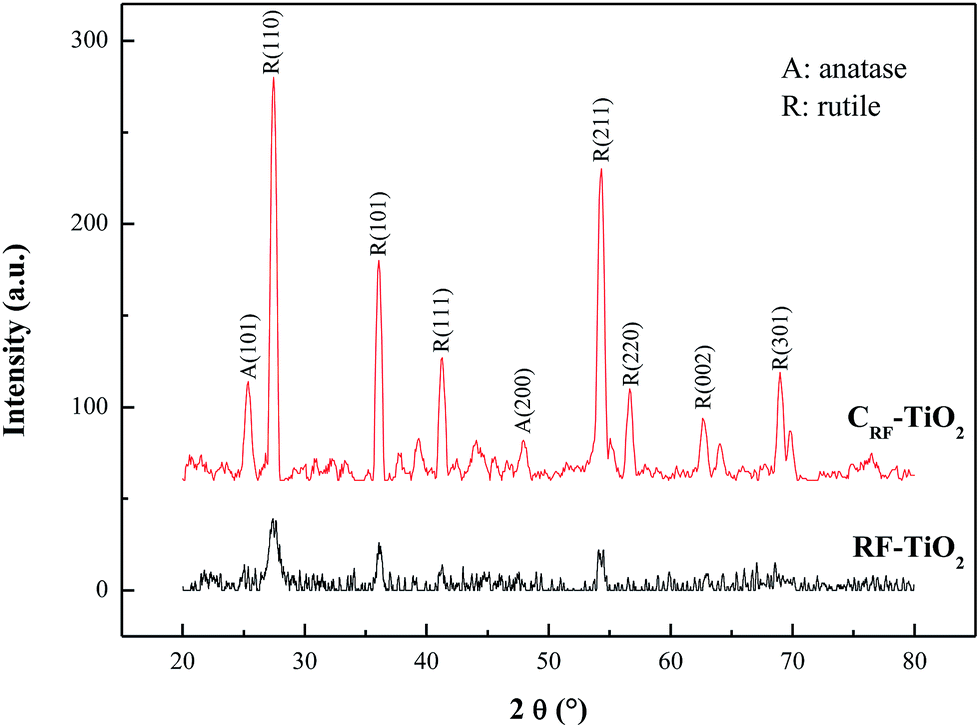
The crystal phase of the fabricated CRF-TiO2 was examined by X-ray diffraction, with RF–TiO2 as comparison. Their XRD patterns are shown in Fig. 2. It can be clearly seen that, above 105 °C dry, weak but identifiable rutile phase with diffraction peaks corresponding to JCPDS no. 21-1276 already existed in the sample, which implies that the rutile TiO2 nanostructures had begun to grow in the RF polymerization process. Strong acidic conditions with the presence of abundant Cl−, have been reported to favor the formation of rutile phase,26 and solution conditions in the RF + TiCl3 route met both of the requirements exactly, thus yielded rutile TiO2. In addition, it is worth noting that, after 500 °C calcination, the crystallinity of TiO2 was greatly enhanced, and a certain amount of anatase TiO2 (JCPDS no. 21-1272) appeared along with the rutile phase, with anatase![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :rutile ratio = 17:83, estimated from XRD intensity data by using the formula expressed as follows:
:rutile ratio = 17:83, estimated from XRD intensity data by using the formula expressed as follows:| | |
χR = (1 + 0.8IA/IR)−1
| (2) |
where χR is the rutile phase ratio, and IA and IR are the integrated intensities of the anatase (101) and rutile (110) diffraction peaks, respectively.27 Anatase–rutile mixture has been reported to be beneficial for effective carrier separation under irradiation, reducing the electrons and holes recombination, and leading to high photocatalytic activity.28 Considering the enhanced crystallinity after calcination with anatase–rutile mixture, CRF-TiO2 is supposed to possess good photocatalytic activity.
 |
| | Fig. 2 XRD patterns of CRF-TiO2 and RF–TiO2. | |
FESEM was employed to observe the morphology of CRF-TiO2 with RF–TiO2 as a comparison to illustrate the growth pathway of CRF-TiO2 during the RF + TiCl3 process. Fig. 3a clearly shows that uniform RF spheres with diameter of around 2–3 μm were dominant in the RF–TiO2 sample, and TiO2 was the minor species, present in a small aggregate state and in close contact with the RF spheres. XRD analysis revealed that the crystallinity of TiO2 was enhanced after 500 °C calcination, and this is consistent with the FESEM observations, as clearly shown in Fig. 3b, where TiO2 became the dominant species in the CRF-TiO2 sample, since part of the RF spheres was decomposed through the loss of CO2, ClOx, H2O etc. during the high temperature calcination,29 and the residuals were carbonized into carbon spheres, evidenced by the elemental mapping images shown in Fig. S1.† The diameter of the carbon spheres was reduced to 1–2 μm, smaller than that of the fresh RF spheres in the RF–TiO2 sample. Fig. 3c shows the higher magnification image of TiO2 as enlarged from the selected area in Fig. 3b. It clearly shows that the TiO2 in the CRF-TiO2 sample had a 3D hierarchical structure with a spherical profile, and was formed by radial growth of elongated crystalline nanorods on the TiO2 nanoparticulate core. This morphology was completely different from the disordered structure of TiO2 produced via direct hydrolysis and oxidation of TiCl3 without the addition of resorcinol and formaldehyde.30 Therefore, it is reasonable to suppose that the nucleation and growth of TiO2 in the RF + TiCl3 process were confined by the RF resin as the template, which restricted the fast and random growth of TiO2 nanoparticles, leading to the formation of ordered, radially aligned TiO2 nanorods inside the RF spheres. After 500 °C calcination, the spherical resin shell decomposed, thus leaving the TiO2 nanorod microsphere structure. The dimension of the TiO2 spheres was around 500–700 nm, and the length and diameter of the TiO2 nanorods were hundreds and tens of nanometers, respectively.
 |
| | Fig. 3 (a) FESEM image of RF–TiO2, (b) FESEM image of CRF-TiO2, (c) enlarged FESEM image of TiO2 in CRF-TiO2, (d) HRTEM image of an individual TiO2 nanorod in CRF-TiO2, inset: SAED pattern of the TiO2 nanorod. | |
Fig. 3d gives the HRTEM image of an individual TiO2 nanorod in the CRF-TiO2 sample. The distance between the adjacent lattice fringes was measured to be 0.325 nm, which agrees well with the interplanar distance of rutile TiO2 (110), indicating the growth direction of [001]. The corresponding selected area electron diffraction (SAED) pattern in the inset of Fig. 3d further solidified the single crystal characteristic of the TiO2 nanorod. It has been well reported that Cl− could preferentially adsorb on the rutile (110) plane and retard the growth rate of the (110) surface, facilitating the anisotropic growth of rutile nanorods along the [001] orientation,26 and this was observed in our study.
An appropriate C/TiO2 ratio is essential for CRF-TiO2 to obtain high pollutant removal capability, since sufficient carbon in the sample can significantly improve the adsorption capability. However, to maintain a high photocatalytic activity, high TiO2 content is also necessary to provide enough active sites exposure for the pollutant photodegradation. In this study, controlled oxidation was performed during the high temperature calcination to adjust the TiO2 content in the final product, and the TiO2 ratio in the CRF-TiO2 sample was 78%, determined by TGA analysis, as shown in Fig. S2.† This ratio is expected to balance the competition between the adsorption capability and the photocatalytic activity.5
Quantitative XPS analysis of CRF-TiO2 was performed to investigate the surface chemical composition and electronic structure of the sample, with survey spectrum and high resolution scans shown in Fig. 4. In this study, we applied peak fitting to the high resolution scan spectra using Gaussian–Lorentzian peak shape after subtraction of the Shirley background. The survey spectrum presented in Fig. 4a confirms the existence of C, O and Ti in the sample, and no peaks corresponding to Cl (e.g. Cl 2p at 198.7 eV) could be found, indicating that Cl was decomposed into gas products during the high temperature calcination process.
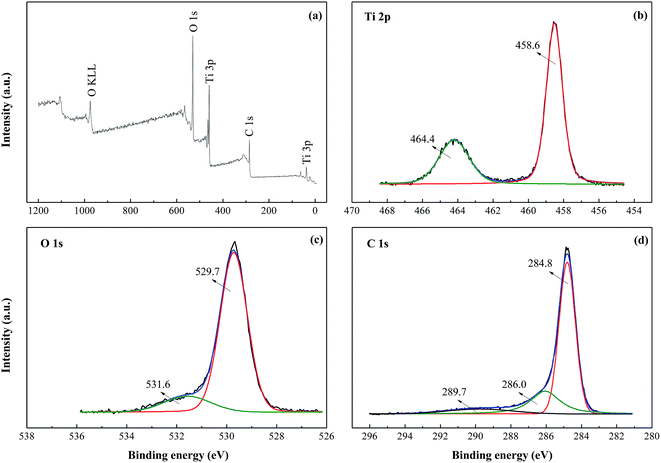 |
| | Fig. 4 (a) XPS survey spectrum of CRF-TiO2 and high-resolution spectra of (b) Ti 2p, (c) O 1s, and (d) C 1s. | |
In the high resolution scan spectrum of Ti 2p (Fig. 4b), binding energies of Ti 2p3/2 and Ti 2p1/2 were found at 458.6 and 464.4 eV, respectively, which is assigned to Ti4+,31 revealing that Ti3+ was oxidized into Ti4+ during the RF + TiCl3 process. The high resolution O 1s spectrum (Fig. 4c) shows two peaks: the prominent peak at about 529.7 eV is associated with oxygen bound to Ti4+ in TiO2, and the broad shoulder centered at 531.6 eV indicates the presence of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O.32 The C 1s spectrum (Fig. 4d) could be deconvoluted into three peaks with binding energies at 284.8 eV, 286.0 eV and 289.7 eV. The strong peak at 284.8 eV is usually assigned to adventitious elemental carbon,33 which is inevitable in XPS measurement; nevertheless, in this study, this peak could also arise from the carbon spheres obviously observed in the FESEM images. Peaks at 286.0 eV and 289.7 eV suggest the existence of –C–O and –CO/O–CO bonds, respectively, which indicates the formation of carbonated species.34 Carbonated species could act as photosensitizers to induce the visible light absorption and response,35 and probably enhance the visible light photocatalytic activity of the CRF-TiO2 sample.33 In addition, a peak corresponding to the Ti–C bond at around 281 eV,31 could not be found, indicating that carbon did not substitute oxygen atoms in the lattice of TiO2.
O.32 The C 1s spectrum (Fig. 4d) could be deconvoluted into three peaks with binding energies at 284.8 eV, 286.0 eV and 289.7 eV. The strong peak at 284.8 eV is usually assigned to adventitious elemental carbon,33 which is inevitable in XPS measurement; nevertheless, in this study, this peak could also arise from the carbon spheres obviously observed in the FESEM images. Peaks at 286.0 eV and 289.7 eV suggest the existence of –C–O and –CO/O–CO bonds, respectively, which indicates the formation of carbonated species.34 Carbonated species could act as photosensitizers to induce the visible light absorption and response,35 and probably enhance the visible light photocatalytic activity of the CRF-TiO2 sample.33 In addition, a peak corresponding to the Ti–C bond at around 281 eV,31 could not be found, indicating that carbon did not substitute oxygen atoms in the lattice of TiO2.
The UV-visible absorption spectrum of CRF-TiO2 was measured in this study, with P25 as reference. Fig. 5a clearly shows that, compared to P25, CRF-TiO2 exhibited strong absorption in the visible light region with long band tailing, but showed no obvious red-shift of absorption threshold. The Kubelka–Munk function was employed to convert the diffuse reflectance spectrum into the equivalent absorption coefficient,36 with the plot of (αhν)1/2 versus hν shown in Fig. 5b. The bandgap of CRF-TiO2 was then calculated to be 3.0 eV, slightly narrower than P25 (3.07 eV). The narrower bandgap of CRF-TiO2 is due to its higher rutile content compared to P25, learned by XRD analysis. Based on theoretical calculation, interstitial carbon in the TiO2 lattice can induce the visible absorption of TiO2,37 while CRF-TiO2 exhibited excellent visible light absorption capability, thus it is reasonable to speculate that a portion of the carbonaceous species in CRF-TiO2, whose existence was proved by XPS analysis, was located at interstitial positions of the TiO2 lattice. In addition, it has been reported that carbon occupying oxygen sites could result in an obvious red-shift of the optical absorption edge;38 however, no obvious red-shift could be found in CRF-TiO2's spectrum, therefore reconfirming that no oxygen atoms were substituted by the incorporated carbon in the sample, which is consistent with the XPS analysis. The strong light absorption of CRF-TiO2 could also be ascribed to its hierarchical structure, which has been reported to be beneficial for efficient photon harvesting due to its excellent light scattering capability.39
 |
| | Fig. 5 (a) UV-visible absorption spectra and (b) Kubelka–Munk transformed reflectance spectra of CRF-TiO2 and P25. | |
The porous structure of the synthesized CRF-TiO2 was examined by N2 adsorption/desorption isotherms, as shown in Fig. 6. It can be seen that the isotherm exhibited a typical type IV curve with a hysteresis loop, indicating the mesoporous structure of CRF-TiO2.40 The inset of Fig. 6 is the pore size distribution curve of CRF-TiO2, as calculated by the BJH and HK method. In this curve, a dominant peak at around 4.7 nm was observed, which reconfirmed the existence of the mesoporous structure, and the mesopores could be ascribed to the interspaces between different TiO2 nanorods inside the hierarchical TiO2. In addition, micropores with pore size around 0.4 nm, corresponding to the pores inside the carbon spheres, could also be clearly observed in the pore size distribution curve. An abundance of meso- and micro-pores usually predicts a large specific surface area for the sample,41 and the BET surface area of the synthesized CRF-TiO2 was measured to be 132.2 m2 g−1 in this study, which is much larger than that of P25.
 |
| | Fig. 6 Isotherm of N2 adsorption/desorption on the surface of CRF-TiO2, inset: pore size distribution of CRF-TiO2. | |
The hierarchical and mesoporous structure, relatively large specific surface area, mature crystallinity with anatase–rutile mixture and enhanced light absorption capability are all important for the adsorption capability and/or photocatalytic activity. Therefore, it is reasonable to suppose that CRF-TiO2 produced via the RF + TiCl3 route possesses excellent pollutant removal capability.
Adsorption capability of CRF-TiO2
A good adsorption capability is essential for rapid pollutant removal and important for a photocatalyst to ensure the subsequent photocatalytic degradation takes place at a high and steady rate. Fig. 7 shows the adsorption of GR on CRF-TiO2 with different initial dye concentrations and contact times, with P25 for comparison. It was found that adsorption of dye on CRF-TiO2 proceeded very quickly; the adsorption amount increased sharply within the initial 20 minutes, and then gradually slowed down until equilibrium was approached. Moreover, under the same initial concentration of 30 mg L−1, the adsorption capacity of CRF-TiO2 was more than 8 times larger than that of P25. A fast adsorption rate and high adsorption capability are both very meaningful for the rapid and effective removal of pollutants, thus, CRF-TiO2 could be a potential candidate for practical pollution remediation. In addition, Fig. 7 also illustrates that, with the increase of initial dye concentration, the adsorption capacity of GR on CRF-TiO2 increased, and a longer time was required to reach equilibrium due to the drastic competition of dye molecules for the limited active sites of the adsorbents.42 In order to understand the dye adsorption behavior and mechanism on the CRF-TiO2 surface, adsorption kinetics and isotherm studies were carried out.
 |
| | Fig. 7 Adsorption kinetics for GR adsorption on CRF-TiO2 and P25, and nonlinear curve-fitting of adsorption on CRF-TiO2 by pseudo-first-order model and pseudo-second-order model. | |
Adsorption kinetics study
In this study, the nonlinear curve-fitting of the pseudo-first-order model (eqn (3)), pseudo-second-order model (eqn (4)) and intra-particle diffusion model (eqn (5)) were applied to analyze the kinetic adsorption behavior.| |
 | (4) |
where qe (mg g−1) and qt (mg g−1) represent the adsorption amount of GR on TiO2 at equilibrium and time t (min), respectively; k1 (min−1), k2 (g mg−1 min−1) and kpi (mg g−1 min−1/2) are the rate constants of pseudo-first-order, pseudo-second-order and intra-particle diffusion model, respectively; and C is a dimensionless constant.
The fitted curves using pseudo-first-order and pseudo-second-order models are shown in Fig. 7, while the intra-particle diffusion model is shown in Fig. S3.† The parameters calculated based on the three equations are listed in Table 1. It could be clearly found that the pseudo-second-order model gave high correlation coefficient values (R2 > 0.99) and exhibited good consistency between the calculated qe and experimental data, which indicates that the pseudo-second-order model could represent the GR adsorption kinetic data well, suggesting that the adsorption may involve the valency forces through sharing or exchange of electrons between the adsorbent and the adsorbate.43 The involvement of a chemical adsorption process could also be deduced from the variations of the O 1s spectrum in XPS analysis implemented on CRF-TiO2 before and after the adsorption, as shown in Fig. S4.† After adsorption, the binding energy of the O 1s peak corresponding to –CO decreased from 531.6 eV to 530.4 eV, which indicated that the electron cloud density of O atom increased, suggesting that the O atom accepted electrons during the adsorption.44 –OH groups in the GR molecule could serve as electron donors. Therefore, combined with the XPS analysis, it is reasonable to infer that a certain amount of electron donation and acceptance occurred during the GR adsorption on the CRF-TiO2 surface.45
Table 1 Adsorption kinetic parameters for the adsorption of GR on CRF-TiO2
| C0 (mg L−1) |
10 mg L−1 |
30 mg L−1 |
50 mg L−1 |
| qe (exp) (mg g−1) |
20.52 |
44.17 |
51.21 |
|
| First-order kinetics |
| k1 (min−1) |
0.0294 |
0.0220 |
0.0210 |
| qe (cal) (mg g−1) |
13.58 |
31.29 |
40.60 |
| R2 |
0.9734 |
0.9575 |
0.9734 |
|
| Second-order kinetics |
| k2 (g mg−1 min−1) |
8.187 × 10−3 |
2.681 × 10−3 |
1.637 × 10−3 |
| qe (cal) (mg g−1) |
20.96 |
45.05 |
52.91 |
| R2 |
0.9993 |
0.9979 |
0.9974 |
|
| Intra-particle diffusion |
| Kp1 (mg g−1 min−1/2) |
4.5579 |
9.0256 |
7.5358 |
| C |
0.0051 |
0.8167 |
0.2234 |
| R2 |
0.9978 |
0.9824 |
0.9937 |
| Kp2 (mg g−1 min−1/2) |
0.9833 |
2.0666 |
2.8591 |
| C |
9.9715 |
19.1331 |
16.9315 |
| R2 |
0.9619 |
0.9841 |
0.9804 |
| Kp3 (mg g−1 min−1/2) |
— |
0.3076 |
0.4039 |
| C |
20.5172 |
39.0101 |
44.4172 |
| R2 |
0.9999 |
0.8282 |
0.8518 |
In addition, the intercept C in the intra-particle diffusion model was not zero, implying that besides the intra-particle diffusion, the boundary layer diffusion may also be the rate-limiting step,46 especially when the initial dye concentration was high.
Adsorption isotherm study
Adsorption isotherm analysis is very important to elaborate the mechanism of an adsorption process. In this study, Langmuir, Freundlich and Dubinin–Radushkevich isotherm models were employed to fit the isotherm data. The Langmuir isotherm model (eqn (6)–(8)) assumes that the monolayer adsorption takes place on a homogeneous surface with equivalent adsorption sites, the Freundlich model (eqn (9)) assumes multilayer adsorption occurs on a heterogeneous surface with non-uniform adsorption sites, and the Dubinin–Radushkevich model (eqn (10) and (11)) can be applied in both heterogeneous and homogeneous systems.| |
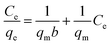 | (6) |
| | |
lgqe = lgKF + (1/n)lgCe
| (8) |
| | |
lnqe = lnqs − kadε2
| (9) |
| | |
ε = RTln(1 + Ce−1)
| (10) |
where C0 and Ce (mg L−1) are the GR concentrations at the beginning and equilibrium, respectively; b (L mg−1), KF (mg g−1 (1/mg1/n)−1) and kad (mol2 kJ−2) are the Langmuir, Freundlich and Dubinin–Radushkevich constants, respectively; qm and qs (mg g−1) are the maximum saturated adsorption capacity and theoretical saturation capacity of the adsorbent, respectively; RL is the characteristic constant of the Langmuir isotherm and is dimensionless; n is the adsorption intensity indicator in the Freundlich model and is dimensionless; R and T are the gas constant (8.314 J mol−1 K−1) and solution temperature (K), respectively.
The isotherm parameters and correlation coefficients were calculated and are presented in Table 2. It can be seen that the adsorption data could be well fitted by the Langmuir equation with high correlation coefficients (R2 > 0.99), implying that homogeneous and monolayer adsorption was predominant and no steric hindrance and lateral interaction occurred between GR and the absorbent.42 The maximum adsorption capacity of GR on CRF-TiO2 deduced from the Langmuir equation was 53.7 mg g−1, which was equivalent and even higher than those reported for some activated carbon,47 suggesting that CRF-TiO2 could be a good absorbent for pollutant removal, and the good adsorption capacity could be attributed to its relatively large specific surface area and mesoporous structure.32 In addition, as shown in Table 2, the calculated RL value was between 0 and 1, and n values in Freundlich model were greater than 5, both of which indicate that the adsorption of dye on CRF-TiO2 was favorable.42 The E of the Dubinin–Radushkevich model is the free energy change of adsorption, giving an indication of the adsorption mechanism. In this study, E was calculated to be between 1–8 kJ mol−1, suggesting that the dominant adsorption mechanism involved was physical adsorption.
Table 2 Isotherm parameters for GR adsorption on CRF-TiO2 at 293 K
| Isotherms |
Parameters |
| Langmuir |
qm (mg g−1) |
53.7634 |
| b (L mg−1) |
0.9163 |
| RL |
0.0135 |
| R2 |
0.9984 |
| Freundlich |
KF (mg g−1 (1/mg1/n)−1) |
26.3027 |
| n |
5.0050 |
| R2 |
0.97 15 |
| Dubinin–Radushkevich |
E (kJ mol−1) |
2.7485 |
| qs (mg g−1) |
45.5101 |
| R2 |
0.8593 |
Photocatalytic activity of CRF-TiO2
Based on the adsorption behavior and mechanism study, we proceeded to investigate the photocatalytic activity of CRF-TiO2 with GR as the probe contaminant. Control experiments illustrated that GR could scarcely be degraded by visible light photolysis alone, but UV light did have a certain ability to decolorize GR without any photocatalyst. After 60 minutes of the UV photolysis process, about 6% of GR could be removed when the C0 of the dye was 50 mg L−1. Nevertheless, compared to the adsorption and photodegradation induced by CRF-TiO2, the amount of GR removed by UV photolysis was negligible.
Photocatalytic activity under UV light irradiation
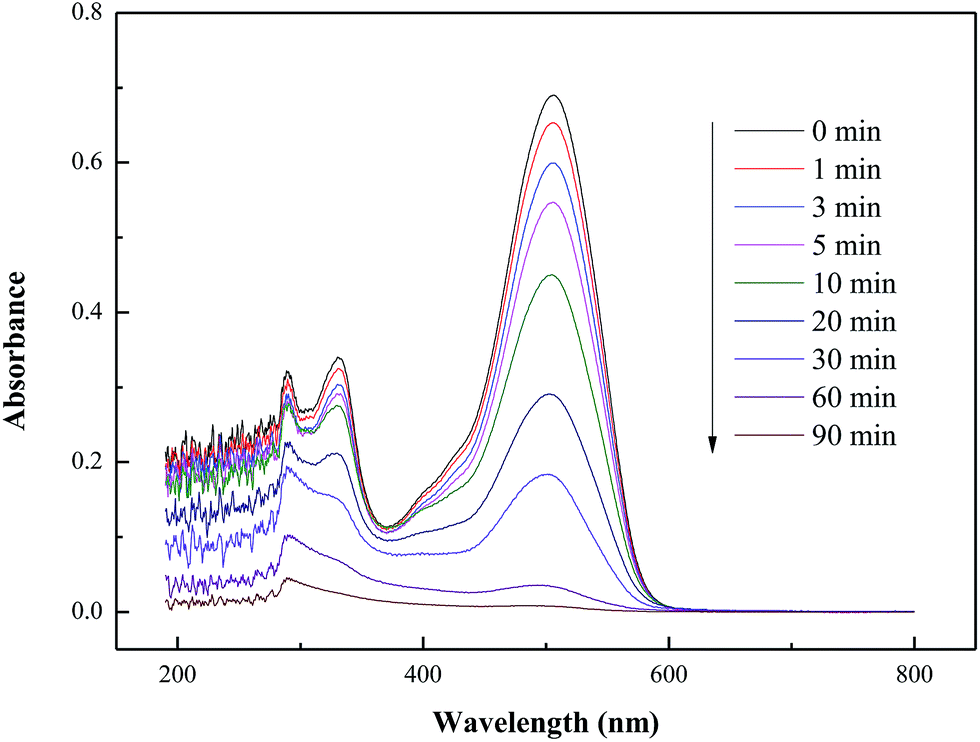
The UV-visible spectra of GR under UV light irradiation in the presence of CRF-TiO2 (Fig. 8) clearly shows that dye removal proceeded very fast, mirrored by the rapid drop in the characteristic peak intensity at 511 nm. Under UV irradiation, photoinduced holes and hydroxyl radicals were generated on the surface of the TiO2 photocatalyst, which could readily oxidize and degrade the GR in the reaction solution.48 After 90 minutes of UV irradiation, the peak at 511 nm faded away without any new band appearing, implying the complete decolorization of the chromophoric groups in the dye. It should be noted that, besides decolorization, mineralization, which refers to the decomposition of dye molecules, is a more essential indicator for effective and complete pollutant removal. In this study, the TOC of the dye solution during the photocatalytic test was also measured to reveal the mineralization extent of GR by CRF-TiO2. It was found that, as shown in Fig. S5,† photocatalytic mineralization of GR proceeded along with, but slower than, decolorization of dye. After 90 minutes of UV irradiation, a TOC loss of approximately 53.2% was attained with CRF-TiO2.
 |
| | Fig. 8 The UV-visible spectra of GR under UV light irradiation with presence of CRF-TiO2. | |
P25 is widely used as a benchmark for new synthesized photocatalysts owing to its good photocatalytic activity. In this study, the pollutant removal capability of CRF-TiO2 was compared with that of P25, with the initial GR concentration before adsorption fixed at 50 mg L−1 for both CRF-TiO2 and P25 systems. It is worth noting that the equilibrium concentrations of GR in the liquid phase before UV irradiation are not the same for CRF-TiO2 and P25 due to the great disparity of their adsorption capability for dye. Therefore, it is difficult to compare solely the effect of the variable photocatalyst due to the fact that changing the dye concentration in the liquid phase also alters the amount of GR adsorbed on the photocatalyst. Therefore, in this study, we simplified the comparison by unifying the initial dye concentrations, rather than the equilibrium concentrations before photocatalysis.
Fig. 9 demonstrates the comparison of GR concentration variation during the integrated adsorption and UV photocatalysis processes for the CRF-TiO2 and P25 systems. The GR concentration here was determined by its absorption at 511 nm. Fig. 9 clearly illustrates that, under UV irradiation, both photocatalysts could catalyze the dye decomposition rapidly, and the exponential decay curves of GR suggest that the decomposition of dye follows pseudo first order reaction kinetics.49 The kinetic rate constants (k, min−1) of the CRF-TiO2 and P25 systems in the first 20 minutes of reaction, calculated by applying the pseudo first order kinetics model, were 0.44 (R2 = 0.993) and 0.50 (R2 = 0.990), respectively, indicating that the photocatalytic activity of CRF-TiO2 was slightly lower than that of P25. However, compared to P25, the excellent adsorption capability of CRF-TiO2 could significantly depress the aqueous GR concentration for photocatalysis, leading to a shorter time requirement (90 minutes for CRF-TiO2 vs. 120 minutes for P25) for the complete decolorization of GR with the same initial concentrations (50 mg L−1). Therefore, compared to P25, CRF-TiO2 is a more attractive photocatalyst for pollutant removal under UV light irradiation.
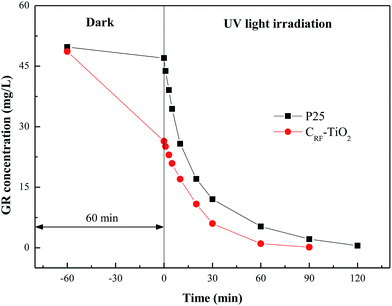 |
| | Fig. 9 Concentration profile of GR in the dark period and in the UV irradiation period with presence of CRF-TiO2 and P25. | |
Photocatalytic activity under visible light irradiation
Enhanced visible light absorption capability and carbon incorporation, confirmed by sample characterization, signify that CRF-TiO2 might possess photocatalytic activity even under visible light irradiation.14,34 As shown in Fig. 10, under the same reaction conditions with a short-arc xenon lamp as light source, P25, which was hardly excited by visible light, showed no photocatalytic activity; nevertheless, CRF-TiO2 could catalyze dye degradation at a certain speed.
 |
| | Fig. 10 Concentration profile of GR in the dark period and in the visible light irradiation period with presence of CRF-TiO2 and P25. | |
The dimensions of the fabricated CRF-TiO2 were on the micrometer scale. After pollutant removal, the photocatalyst could be easily recovered by gravity sedimentation or membrane filtration without causing serious membrane fouling. This part of the experiment along with the study of reusability CRF-TiO2 by our group is currently being done by our group and will be reported later. Neglecting the advantage of easy recovery and reusability, the excellent adsorption capability and photocatalytic activity under both UV and visible light irradiation are fully capable of illustrating that CRF-TiO2 is a good candidate for practical water purification.
Conclusion
In summary, a carbon incorporated hierarchical TiO2 nanostructure (CRF-TiO2) was successfully synthesized via a facile one-pot route and was well characterized in this study, and its pollutant removal capabilities including adsorption and photocatalysis, were fully investigated. It was found that the synthesized CRF-TiO2 possessed a relatively large specific surface area with an interesting hierarchical and mesoporous structure, leading to an excellent pollutant adsorption capability, where the equilibrium adsorption capacity of CRF-TiO2 was more than 8 times larger than that of Degussa P25, and the adsorption kinetics and isotherm studies revealed that pseudo-second-order model and Langmuir isotherm model could successfully fit the adsorption data. Furthermore, CRF-TiO2 possessed excellent light absorption capability, and TiO2 in the final product was an anatase–rutile mixture with mature crystallinity, all of which equipped CRF-TiO2 with excellent photocatalytic activity under both UV and visible light irradiation. Compared to the benchmark P25, CRF-TiO2 could remove pollutants in a much faster and more efficient manner. In addition, considering the mass production feasibility of the RF + TiCl3 route, CRF-TiO2 has great potential in practical pollutant removal applications.
Acknowledgements
This work was supported by the Research Foundation for the Outstanding Young and Middle-aged Scientists of Shandong Province of China (BS2014HZ005), China Postdoctoral Science Foundation (2013M540550, 2014T70643), Tai Shan Scholar Foundation (TS201511003), and Shanghai Tongji Gao Tingyao Environmental Science & Technology Development Foundation.
References
- Y. Ohko, I. Ando, C. Niwa, T. Tatsuma, T. Yamamura, T. Nakashima, Y. Kubota and A. Fujishima, Environ. Sci. Technol., 2001, 35, 2365–2368 CrossRef CAS PubMed.
- A. Fujishima, X. T. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- R. Sedghi and F. Heidari, RSC Adv., 2016, 6, 49459–49468 RSC.
- J. H. Pan, X. Z. Wang, Q. Z. Huang, C. Shen, Z. Y. Koh, Q. Wang, A. Engel and D. W. Bahnemann, Adv. Funct. Mater., 2014, 24, 95–104 CrossRef CAS.
- R. Leary and A. Westwood, Carbon, 2011, 49, 741–772 CrossRef CAS.
- J. H. Pan, G. Han, R. Zhou and X. S. Zhao, Chem. Commun., 2011, 47, 6942–6944 RSC.
- I. Tamiolakis, I. T. Papadas, K. C. Spyridopoulos and G. S. Armatas, RSC Adv., 2016, 6, 54848–54855 RSC.
- G. Wang, W. J. Feng, X. K. Zeng, Z. Y. Wang, C. P. Feng, D. T. McCarthy, A. Deletic and X. W. Zhang, Water Res., 2016, 94, 363–370 CrossRef CAS PubMed.
- J. J. Lin, Y. U. Heo, A. Nattestad, Z. Q. Sun, L. Z. Wang, J. H. Kim and S. X. Dou, Sci. Rep., 2014, 4, 5769 CrossRef PubMed.
- L. Wang, Z. Y. Nie, C. B. Cao, Y. Q. Zhu and S. Khalid, J. Mater. Chem. A, 2015, 3, 6402–6407 CAS.
- H. W. Bai, L. Liu, Z. Y. Liu and D. D. Sun, Water Res., 2013, 47, 4126–4138 CrossRef CAS PubMed.
- J. C. Liu, W. Y. Zhu, S. Y. Yu and X. L. Yan, Carbon, 2014, 79, 369–379 CrossRef CAS.
- H. W. Bai, Z. Y. Liu and D. D. Sun, J. Mater. Chem., 2012, 22, 18801–18807 RSC.
- M. Q. Yang, N. Zhang and Y. J. Xu, ACS Appl. Mater. Interfaces, 2013, 5, 1156–1164 CAS.
- D. L. Zhao, X. Yang, C. L. Chen and X. K. Wang, J. Colloid Interface Sci., 2013, 398, 234–239 CrossRef CAS PubMed.
- B. Chai, T. Y. Peng, J. Mao, K. Li and L. Zan, Phys. Chem. Chem. Phys., 2012, 14, 16745–16752 RSC.
- Z. H. Zhang, Y. Xu, X. P. Ma, F. Y. Li, D. N. Liu, Z. L. Chen, F. Q. Zhang and D. D. Dionysiou, J. Hazard. Mater., 2012, 209, 271–277 CrossRef PubMed.
- Z. K. Jin, M. Yang, J. J. Wang, H. Y. Gao, Y. F. Lu and G. Wang, Chem.–Eur. J., 2016, 22, 6031–6036 CrossRef CAS PubMed.
- C. X. Zhao, Z. S. Wang, W. Chen, Y. B. Song, X. H. Chen and T. Xie, Funct. Mater. Lett., 2016, 9, 1650031 CrossRef CAS.
- K. S. Park, K. M. Min, Y. H. Jin, S. D. Seo, G. H. Lee, H. W. Shim and D. W. Kim, J. Mater. Chem., 2012, 22, 15981–15986 RSC.
- N. P. Wickramaratne and M. Jaroniec, J. Colloid Interface Sci., 2015, 449, 297–303 CrossRef CAS PubMed.
- M. Lucic, N. Milosavljevic, M. Radetic, Z. Saponjic, M. Radoicic and M. K. Krusic, Sep. Purif. Technol., 2014, 122, 206–216 CrossRef CAS.
- I. K. Konstantinou and T. A. Albanis, Appl. Catal., B, 2004, 49, 1–14 CrossRef CAS.
- T. A. Egerton and H. Purnama, Dyes Pigm., 2014, 101, 280–285 CrossRef CAS.
- S. Ahmed, M. G. Rasul, W. N. Martens, R. Brown and M. A. Hashib, Desalination, 2010, 261, 3–18 CrossRef CAS.
- L. B. Yu, Z. Li, Y. B. Liu, F. Cheng and S. Q. Sun, J. Power Sources, 2014, 270, 42–52 CrossRef CAS.
- H. B. Li, X. C. Duan, G. C. Liu, X. B. Jia and X. Q. Liu, Mater. Lett., 2008, 62, 4035–4037 CrossRef CAS.
- D. C. Hurum, A. G. Agrios, K. A. Gray, T. Rajh and M. C. Thurnauer, J. Phys. Chem. B, 2003, 107, 4545–4549 CrossRef CAS.
- W. M. Zhang, J. S. Hu, Y. G. Guo, S. F. Zheng, L. S. Zhong, W. G. Song and L. J. Wan, Adv. Mater., 2008, 20, 1160–1165 CrossRef CAS.
- E. Hosono, S. Fujihara, K. Kakiuchi and H. Imai, J. Am. Chem. Soc., 2004, 126, 7790–7791 CrossRef CAS PubMed.
- D. E. Gu, Y. Lu, B. C. Yang and Y. D. Hu, Chem. Commun., 2008, 2453–2455 RSC.
- Y. Gao, Q. Y. Yue, B. Y. Gao, Y. Y. Sun, W. Y. Wang, Q. Li and Y. Wang, Chem. Eng. J., 2013, 232, 582–590 CrossRef CAS.
- P. H. Wang, P. S. Yap and T. T. Lim, Appl. Catal., A, 2011, 399, 252–261 CrossRef CAS.
- D. M. Chen, Z. Y. Jiang, J. Q. Geng, Q. Wang and D. Yang, Ind. Eng. Chem. Res., 2007, 46, 2741–2746 CrossRef CAS.
- P. Gorska, A. Zaleska, E. Kowalska, T. Klimczuk, J. W. Sobczak, E. Skwarek, W. Janusz and J. Hupka, Appl. Catal., B, 2008, 84, 440–447 CrossRef CAS.
- K. Sunada, T. Watanabe and K. Hashimoto, Environ. Sci. Technol., 2003, 37, 4785–4789 CrossRef CAS PubMed.
- H. Kamisaka, T. Adachi and K. Yamashita, J. Chem. Phys., 2005, 123, 84704–84709 CrossRef PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- Y. C. Rui, Y. G. Li, Q. H. Zhang and H. Z. Wang, Nanoscale, 2013, 5, 12574–12581 RSC.
- S. P. Xu, A. J. Du, J. C. Liu, J. Ng and D. D. Sun, Int. J. Hydrogen Energy, 2011, 36, 6560–6568 CrossRef CAS.
- Y. Gao, Q. Y. Yue, S. P. Xu, B. Y. Gao and H. Yu, Chem. Eng. J., 2015, 274, 76–83 CrossRef CAS.
- Y. Gao, Q. Y. Yue, B. Y. Gao and Y. Y. Sun, Desalin. Water Treat., 2015, 55, 624–636 CrossRef CAS.
- K. Y. Foo and B. H. Hameed, Bioresour. Technol., 2012, 104, 679–686 CrossRef CAS PubMed.
- S. X. Teng, S. G. Wang, X. W. Liu, W. X. Gong, X. F. Sun, J. J. Cui and B. Y. Gao, Colloids Surf., A, 2009, 340, 86–92 CrossRef CAS.
- P. Georgiou, J. Walton and J. Simitzis, Electrochim. Acta, 2010, 55, 1207–1216 CrossRef CAS.
- F. Ahmad, W. Daud, M. A. Ahmad and R. Radzi, Chem. Eng. Res. Des., 2012, 90, 1480–1490 CrossRef CAS.
- Y. Gao, Q. Y. Yue and B. Y. Gao, J. Environ. Sci., 2016, 41, 235–243 CrossRef PubMed.
- E. Topkaya, M. Konyar, H. C. Yatmaz and K. Ozturk, J. Colloid Interface Sci., 2014, 430, 6–11 CrossRef CAS PubMed.
- D. Nasuhoglu, V. Yargeau and D. Berk, J. Hazard. Mater., 2011, 186, 67–75 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra20188a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.