
Solution-grown GeO2 nanoparticles with a nearly 100% yield as lithium-ion battery anodes†
Guo-An Li,
Wei-Chin Li,
Wei-Chung Chang and
Hsing-Yu Tuan*
Department of Chemical Engineering, National Tsing Hua University, 101, Section 2, Kuang-Fu Road, Hsinchu, Taiwan 30013, Republic of China. E-mail: hytuan@che.nthu.edu.tw; Fax: +886-3-571-5408; Tel: +886-3-572-3661
First published on 10th October 2016
Abstract
Germanium oxide (GeO2) nanoparticles were synthesized with a nearly 100% production yield in a nonionic reverse micelle system at ambient temperature. The procedure is a facile and energy saving strategy for producing germanium oxide nanoparticles with ultra large throughput. As-prepared GeO2 nanoparticles can be directly used as anode materials without any post-treatment or other supplementary additives for lithium ion batteries. GeO2-anodes exhibited good electrochemical performance in terms of both gravimetric and volumetric capacity. The GeO2 anodes have a reversible capacity of approximately 1050 mA h g−1 at a rate of 0.1C, close to its theoretical capacity (1100 mA h g−1), and good rate capability without severe capacity decade. The volumetric capacity of the GeO2 anodes reaches 660 mA h cm−3, which is higher than the performance of commercial graphite anode (370–500 mA h cm−3). Coin type and pouch type full cells assembled for electronic devices applications were also demonstrated. A single battery is shown to power LED array over 120 bulbs with a driving current of 650 mA. Based on the above, the micelle process of GeO2 nanoparticle synthesis provides a possible solution to high-capacity nanoparticles' scalable manufacturing for lithium ion battery applications.
Introduction
Lithium-ion batteries (LIBs), as a state-of-the-art technology in energy storage devices, have been utilized in a wide range of applications due to their relatively high energy density, long cycle life span, and good rate capability, etc.1–4 Ge-based materials have been intensively studied due to their remarkable high capacities over 1000 mA h g−1 as anode materials for LIBs owing to Li–Ge alloying mechanism.5–8 For example, germanium has a high capacity of 1384 mA h g−1, which is much higher than the theoretical capacity of commercial graphite (372 mA h g−1).9,10 Nevertheless, two challenging problems of Ge-based material must be addressed. One is Ge's huge volume change (∼370%) during charging and discharging procedures which induces active material cracking and fracture and produces fresh surface that consumes lithium and causes irreversibility capacity loss. Recent development of a variety of Ge nanostructures is a highly effective strategy to improve the negative influence of electrode's volume expansion on battery performance.11–15 On the other hand, the other critical problem is that the limitation of Ge's high cost and expensive manufacturing process, but this issue is rarely addressed.16Germanium dioxide, also called germania, is much less expensive than germanium, and widely used in optical fibers, polymer catalysts, and also as a component of waveguides where it is capability of modulating the index of refraction.17,18 In general, various metal oxide nanostructures exhibit excellent electrochemical performance in lithium-ion batteries.19–22 GeO2 reacts with lithium ion and transforms irreversibly into germanium nanoparticles and Li2O matrix during beginning of reduction process, and then germanium nanoparticles react reversibly with lithium ion through alloying mechanism for the following lithiation/delithiation process.23,24
| GeO2 + 4Li → Ge + 2Li2O |
| Ge + 4.4Li ↔ Li4.4Ge |
Based on the above reaction, GeO2 has a theoretical capacity of 1100 mA h g−1 (4653 mA h cm−3) which is comparable to germanium's performance. However, the practical capacity of GeO2 is not well-maintained. The rapid capacity loss of the GeO2 electrode fabricated by Brousse et al.25 dropped from 740 to 225 mA h g−1 after 10 cycles is attributed to the ∼230% volume change during electrochemical test. Recently, lots of efforts have been made to address situations to obtain progressive improvement. GeO2 electrode maintains cycling stability with the aid of carbonaceous additives to form composites, which effectively alleviate the stress from volume variation during lithium intake/removal.26–30 In the meanwhile, supplementary additives offer additional electron transfer pathway, which represent kinetics improvement related to rate capability for LIBs. Another strategy to ameliorate GeO2 poor performance is size-controlled and architecture-design of the active material. Size and morphology of active material indeed play an important role in electrochemical performance for LIBs. Various nanostructures of GeO2, such as nanotube,31 nanoparticle,32,33 3-D porous nanostructure,34 exhibit a high reversible capacity and capacity retention with slightly capacity fade, which implies the alleviation of stress from Li intercalation/de-intercalation to maintain electrode integrity. They also perform well in rate capability determined by the kinetics of electron conductivity and lithium diffusivity. On the basis of time constant τ, kinetics strongly depend on the diffusivity length (τ ≈ L2D−1). However, these methods used to synthesize GeO2 nanostructures, such as thermal oxidation,35,36 template-directed method,37–39 and thermal evaporation,40 usually need harsh experimental conditions or complicated manufacturing process, which not only produce unnecessary impurities harmful to environment but also increase the manufacturing cost. Those synthetic methods for GeO2 might not be suited for industrial-scale production. Besides, GeO2 nanomaterials were usually incorporated with carbon additives, such as carbon nanotube, graphene, 2-D matrix for assembling into LIBs. The batteries gain stable cycling life or increase specific capacity, but sacrifice the overall energy density due to the addition of extra volume and weight to electrode. Therefore, a suitable strategy is required to fabricate GeO2 nanoparticles on the purpose of sustainability and scalability.
Herein, we report the synthesis of high quality hexabranched GeO2 nanoparticles at room temperature with a nearly 100% yield via an optimized sol–gel process (reverse micelle). With well controlling the oil/water phase, surfactant/co-surfactant ratio and choosing the right germanium source, the well-defined GeO2 nanoparticles can be obtained abundantly. In addition, as-prepared GeO2 nanoparticles were used as anode materials for lithium ion batteries without any post-treatment. The electrochemical results demonstrate high capacity and quite stable capacity retention. Finally, coin type and pouch type full cells assembled with the GeO2 as anode and Li(NiCoMn)O2 as cathode are fabricated and evaluated. Pouch type full cells were utilized to power light-emitting-diodes (LEDs) over 120 bulbs with a large current (∼650 mA).
Experimental
Material synthesis
Hexabranched GeO2 nanoparticles were synthesized by a micro emulsion method in a nonionic reverse micelle system at ambient temperature.41 The reverse micelle solution is prepared by well mixing 50 ml hexane (97.05%, Alfa Aesar), 47.4 g Triton-X-100 (Laboratory grade, Sigma Aldrich), 45 ml 1-hexanol (reagent, 98%, Sigma Aldrich), 18 ml pH = 1 hydrogen chloride (HCl) (37%, Sigma Aldrich) solution together first by closing the valve below the reactor, and the mixture is stirred for half an hour in the reactor at room temperature until the solution become transparent. Next, 4.2 g germanium ethoxide (Ge(OEt)4) (99.995%, Alfa Aesar) was injected into the reactor for the further reaction. After about 5 minutes, the resulting solution became turbid under vigorous stirring for one hour at room temperature. White product powders weight in proximity of 1.2 g can be collected on a filter paper.Characterization
To comprehend crystal structure of the product, X-ray diffraction with Cu Kα radiation operated at 40 kV and 20 mA was utilized. The morphology of GeO2 was characterized by field emission scanning electron microscope (FE-SEM, Hitachi SU8010) operated at 10 kV accelerating voltage with working distances ranging between 10 to 20 mm and transmission electron microscope (TEM, JEOL JEM-2100F) equipped with an EDS spectrometer. X-ray photoelectron spectrometer (XPS, PHI Quantera SXM/Auger: AES 650) analysis was executed to figure out the reaction mechanism of GeO2 nanoparticles with lithium metal. The XPS sample was prepared by dissembling GeO2-based coin cell after 100 times charging/discharging test, and the electrode was dispersed in diethyl carbonate (DEC) solvent with ultrasonic treatment half an hour to remove residual electrolyte.Electrochemical characterization
GeO2 nanoparticles were well mixed with conductive material Super P carbon black and PAA binder in ethanol solvent at the weight ratio of 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:20 and then the homogeneous slurry were tape casted onto a copper foil by doctor-blade film coater. The electrodes were dried at 50 °C in vacuum oven to evaporate ethanol solvent and pressed densely for fear of material exfoliation by means of rolling machine. Before cell assembling in Ar-filled glove box, the weight of pure active material is accurately acquired utilizing a microbalance with 0.1 μg resolution (Sartorius SE2) for capacity calculation, giving typical mass loading of 0.6 mg cm−2 The coin-type half-cells (CR2032) were assembled with anode electrodes made of GeO2 nanoparticles, Li metal cathode, and separator soaked in electrolyte. The electrolyte was 1 M LiPF6 in fluoro-ethylene carbonate/diethyl carbonate (FEC/DEC) (3:7 vol%).
:20:20 and then the homogeneous slurry were tape casted onto a copper foil by doctor-blade film coater. The electrodes were dried at 50 °C in vacuum oven to evaporate ethanol solvent and pressed densely for fear of material exfoliation by means of rolling machine. Before cell assembling in Ar-filled glove box, the weight of pure active material is accurately acquired utilizing a microbalance with 0.1 μg resolution (Sartorius SE2) for capacity calculation, giving typical mass loading of 0.6 mg cm−2 The coin-type half-cells (CR2032) were assembled with anode electrodes made of GeO2 nanoparticles, Li metal cathode, and separator soaked in electrolyte. The electrolyte was 1 M LiPF6 in fluoro-ethylene carbonate/diethyl carbonate (FEC/DEC) (3:7 vol%).
In the fabrication of coin-type and pouch-type full cells, the Li(NiCoMn)O2 electrode with a loading mass of 21.12 mg cm−2 was to replace lithium metal as reference/counter electrode, and the anodes GeO2 nanoparticles remained the same loading mass. For perfect full cell assembly, the electrochemical performance of the cathode Li(NiCoMn)O2 was evaluated by half-cell measurement. The slurry for cathode electrode was prepared by mixing active materials (Li(NiCoMn)O2, 94.5 wt%) with 3.5 wt% of Super-P and 2 wt% of PVDF (polyvinylidene fluoride) binder dispersed in NMP (N-methyl-2-pyrrolidone) solvent. From the Fig. S7,† the areal capacity for the cathode electrode was ∼3.3 mA h cm−2 and the working potential of Li(NiCoMn)O2 vs. Li was observed at 3.8 V. For pouch type full cell assembly, GeO2 anode, membrane, and Li(NiCoMn)O2 cathode were stacked orderly in the Al-laminated film after welding metallic strip terminal and then filled with electrolyte. For the sake of electrolyte soaking entirely, pouch type full cell were rest for half hour after cell sealing. All the electrochemical performance of the GeO2-based lithium ion batteries were evaluated using Maccor Series 4000 instruments.
Results and discussion
The micelle system for production of well-defined GeO2 nanoparticles was conducted with the experimental setup represented in the Fig. 1. This process can be easily scaled up if sufficient precursor and suitable reactor volume are provided. Germanium ethoxide (Ge(OEt)4) as organometallic precursor was directly injected into a flask containing hexane, 1-hexanol, Trion-X-100, and HCl solution, which serve as oil phase, surfactant, co-surfactant, and aqueous phase, respectively. (Ge(OEt)4) dissolved in the nano-sized micelle capped with Triton-X-100 and 1-hexanol, and a perfectly mixing process lasted for 60 min. The white powder was obtained and the product GeO2 weighted nearly 1.2 g in one batch shown in the Fig. 2(a). In a typical synthesis, the synthetic method could attain the product yield of approaching 100% under appropriate reaction operation. The large quantity GeO2 nanoparticles obtained from the micelles system were directly used and suitable with current slurry coating technique for fabrication of lithium-ion batteries electrode.42 To comprehend the growth mechanism of well-faceted GeO2 nanoparticles, products formed with different reaction times were evaluated (Fig. S1†). With a reaction time 5 min, the product obtained was aggregated by irregularly shape nanoparticles, indicating a rapidly nucleation and multipoint growth mechanism for formation of GeO2. After 10 min, GeO2 particles have developed into hexabranched nanoparticles with more than 100 nm in length and large size distribution. GeO2 nanoparticles transformed into hexagonally symmetrical shape probably related to its hexagonal crystal structure. After a half hour of reaction, the GeO2 nanoparticles became more uniform in size as the same as those shown in Fig. 1. | ||
| Fig. 1 The illustration of experimental design of GeO2 nanoparticles synthesized in microemulsion system at room temperature. | ||
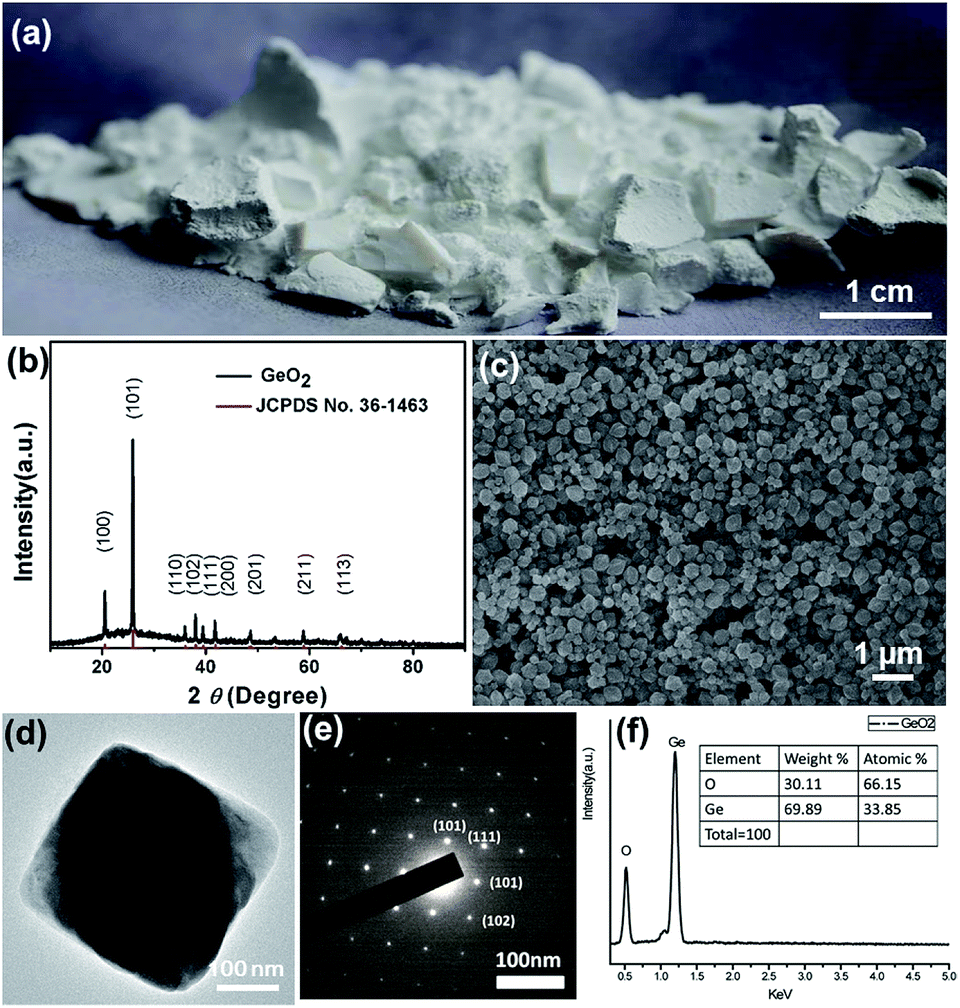 | ||
| Fig. 2 (a) Photographical representation of the 1.2 grams GeO2 powders synthesized by a microemulsion approach at room temperature. (b) XRD pattern, (c) SEM images, (d) TEM images, (e) SAED pattern, and (f) EDS analysis of hexabranched GeO2 nanoparticles. | ||
The crystal structure of GeO2 nanoparticles was investigated by X-ray diffraction analysis, as shown in Fig. 2(b). The XRD pattern perfectly matches the pure hexagonal phase structure of α-GeO2 (JCPDS no. 36-1463 with unit cell constants a = 4.985 Å and c = 5.648 Å), and it is obvious that no impurity phase was observed. Further surface and morphology characterization of GeO2 are provided by SEM images and the shape of product developed in this scale-up experiment design is hexabranched without transformation (Fig. 2(c)). The average length and width of the GeO2 nanoparticles are determined to 200 ± 20 nm and 150 ± 25 nm based on statistics analysis over 500 nanoparticles measured from SEM images. TEM image and its corresponding selected area electron diffraction (SAED) of well-defined GeO2 nanoparticle are given (Fig. 2(d) and (e)). Because of the inherent structural growth, the (101), (111) and (102) planes can be determined by diffraction perpendicular to the long axis. In addition, EDX analysis revealed the composition of GeO2 nanoparticles showing exact atomic ratio shown in Fig. 2(f).
To evaluate the electrochemical performance of GeO2 nanoparticles as anode material for LIBs, the product was assembled into coin type half-cell (CR2032) with Li metal as counter and reference electrode. First of all, the galvanostatic charging/discharging measurement was implemented at the current rate 0.1C (1C = 1.1 A g−1) with working voltage window of 0.01 V–1.5 V at room temperature. The cycling life curve in the Fig. 3(a) reveals that first charge and discharging capacity of the GeO2 nanoparticles are 1948 mA h g−1 and 1074 mA h g−1, respectively, corresponding to the coulombic efficiency of 55.1%. Presumably, the large irreversible capacity loss (874 mA h g−1) in the first cycle is attributed to the formation of Li2O, usually found in other metal oxide species such as silicon oxide.43,44 According to the previous research,23,45–47 it is believed that metal nanoparticles formed in delithiation process have catalytic property to decompose Li2O, which improves the coulombic efficiency of initial charging/discharging test. After the following cycles, the average reversible capacity of GeO2 nanoparticles was 1050 mA h g−1 with the excellent capacity retention of 87% based on the fifth cycle capacity (1105 mA h g−1). The stability of cycling life curve represent the electrode prepared by hexabranched GeO2 nanoparticles could tolerate the stress torture from huge variation during lithium intake and removal. Fig. 3(b) depicted the voltage profiles of the GeO2 anode in the first cycle. During the charge process, the voltage decreases dramatically from the open circuit voltage to approximately 0.7 V, and the plateau region show up, and then the voltage further decreases slowly until around 0.15 V. In the discharge profile, the plateau can be clearly observed from 0.3 V to 0.6 V. To clearly understand the reaction mechanism of GeO2 with lithium, differential capacity profiles shown in the Fig. 4(a) and (b) were derived from the voltage profiles of the first to 100th cycle in Fig. 3(a), and these peaks indicate various electrochemical reactions related to insertion/extraction of Li ions. Observed from the Fig. 4(a), there exist a distinct peak located near 0.5 V in the first charge cycle and vanished in the subsequent cycle, which probably present formation of SEI (solid electrolyte interface) layer and Li2O in irreversibility.33,48 At the following lithiation processes, small peak at approximately 0.13 V correspond to the Li–Ge alloying reaction. Correspondent Li–Ge de-alloying reaction is indexed to the small hump (0.35 V) in the delithiation process. Apparently, over several tens of times cycle test (Fig. 4(b)), the small peak around 0.37 V in the reduction sweep showed up, which is associated with formation of lithium-rich germanium alloys. Furthermore, the peaks at 0.62 V in reduction sweep and 0.7 V in oxidation sweep can be indicated to the redox reaction of germanium. However, the redox reaction reversibility is gradually decreasing, which may be a cause of capacity fading. These results are consistent with those reported research for cyclic voltammetry of GeO2.49,50 In reality, many electronic applications require high current operation. To confirm the feasibility of GeO2 in practical application, high rate test of GeO2-based batteries was executed. As shown in Fig. 5(a), current rate 1C with respect to 1100 mA h g−1 is employed and the discharge capacity is from 982 mA h g−1 in 2nd cycle to 856 mA h g−1 in 50th cycle with 87% capacity retention. Except 1C test, multi-rate with different operating current rates and its voltage profiles are depicted to examine the rate capability of GeO2 anode (Fig. 5(b) and (c)). From Fig. 5(b), the plateau isn't obvious once cycled at high rate, which implies the difficulty in lithium insertion. At a high current density test, the lithium insertion into active material site is rate-determining step, which means the kinetic limitation hinder the further lithium ions react with GeO2 nanoparticles.51,52 The GeO2 anode exhibits discharge capacities of 1250 mA h g−1, 1100 mA h g−1, 1000 mA h g−1, 855 mA h g−1, 590 mA h g−1, 420 mA h g−1, and 250 mA h g−1, corresponding to the various current densities of 0.1C, 0.5C, 1C, 2C, 4C, 6C, and 8C, respectively shown in Fig. 5(c). Finally, the specific discharge capacity of about 1170 mA h g−1 is recovered fast when the rate is returned to 0.1C again after 35 cycles. The additional electrochemical performance of GeO2 nanoparticles are provided in the ESI.† GeO2-based batteries employ different composition electrolytes system showing diverse electrochemical performance. As shown in Fig. S1,† FEC/DMC electrolyte system possesses more stable capacity retention and high rate performance than that of EC/DMC electrolyte system, which is perhaps due to formation of good SEI layer on the electrode surface.53–55
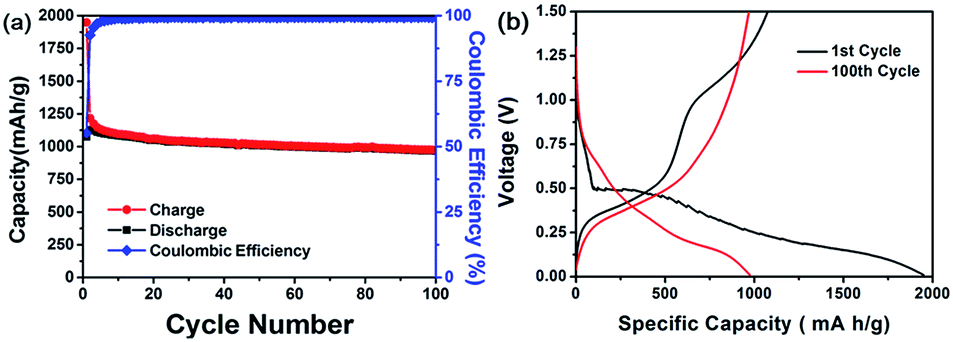 | ||
| Fig. 3 (a) Charge/discharge cycle performance of GeO2 anode at a 0.1C rate between 0.01 V and 1.5 V. (B) Voltage profiles of 1st and 100th cycle at a 0.1C rate. | ||
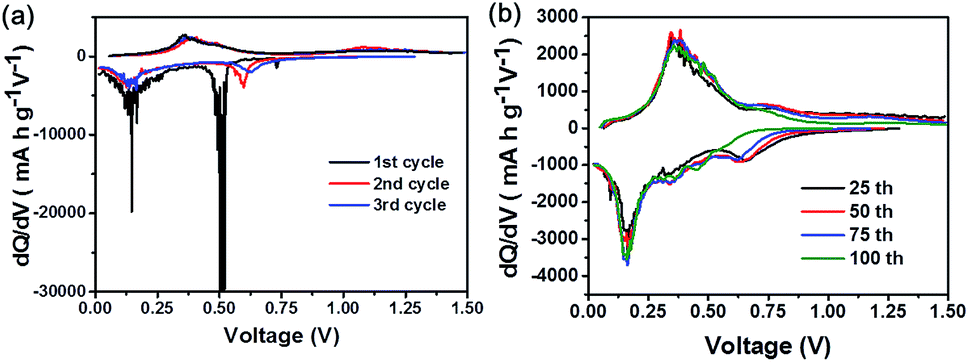 | ||
| Fig. 4 Differential capacity profiles of the GeO2 anode in the (a) 1st, 2nd, 3rd and following (b) 25th, 50th, 75th, 100th cycle derived from the galvanostatic charging/discharging curve at the rate of 0.1C. | ||
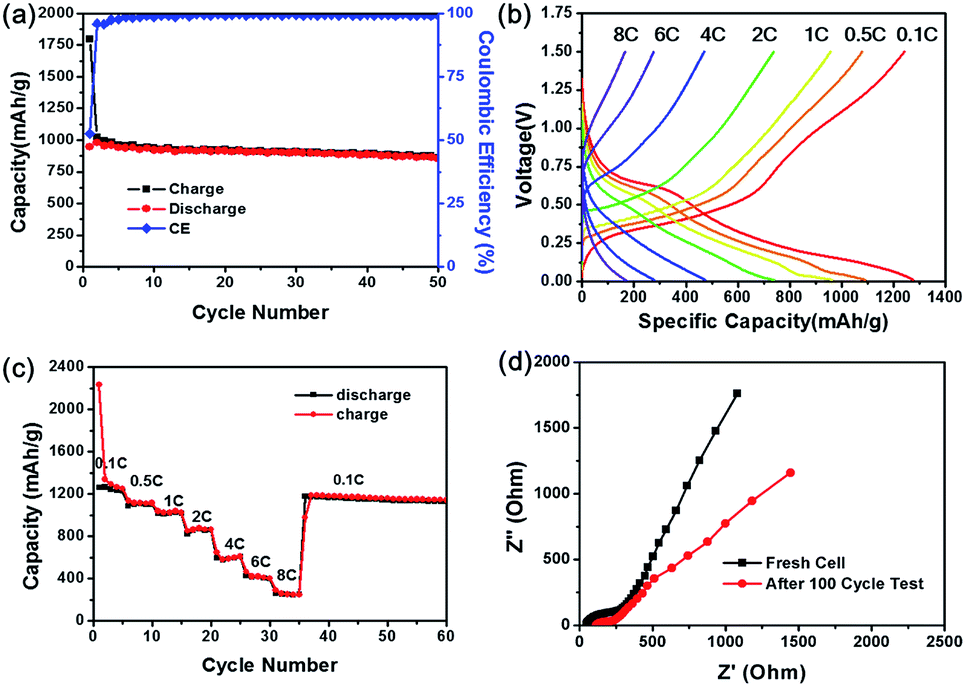 | ||
| Fig. 5 Rate capability of GeO2 nanoparticle anode. (a) Charge/discharge cycle performance of GeO2 anode at a 1C rate between 0.01 V and 1.5 V. (b) Cycle performance of a 50-cycle test at rate range from 0.1C to 8C. (c) Voltage profiles of the performance in a 50-cycle test. (d) Nyquist plot for fresh/after 100 cycle test. | ||
To further understand the interfacial electrochemical behavior of GeO2 electrodes, electrochemical impedance spectroscopy (EIS) analysis was performed at frequencies from 10 kHz to 10 mHz. From the Nyquist plots (Fig. 5(d)), the diameter of semicircle at high frequency region became small after charging/discharging test, which implies that transfer electron resistance reduces possibly due to the composition change of electrode such as formation of Li2O matrix.56–58 As for the inclined straight line at low frequency region, the slope of the line is relevant to lithium ion diffusivity which shows the difficulty for lithium ion immigration after 100 cycles.
In order to know the situation of composition and appearance of GeO2 nanoparticles after electrochemical test, after 100th cycling in galvanostatic charge/discharge cycles at 0.1C rate was disassembled and cleaned with DEC solvent to remove residual byproducts. Fig. 6(a) demonstrates the intuitive evidences for before/after 100 cycling test in coin type cell, from the ex situ XPS measurement (Fig. 6(b)) of GeO2 anode. It is obvious that the peak indicated (32.4 eV) corresponds to the Ge–O bonds. After cycling 100th cycles, the peak representing Ge–O bonding disappear, which is due to the full conversion of GeO2 to form germanium nanoparticles and Li2O. The single peak observed at 29.8 eV is well indicated to the elemental germanium. From XRD patterns shown in Fig. 6(c), there are two obvious peaks indexed to the (100) and (101) crystalline planes of GeO2 nanoparticles with good crystallinity for fresh electrode. After cycled 100 times, the crystalline GeO2 became amorphous, and showed no obvious peak from XRD pattern. Consequently, these evidences demonstrate that active material in the beginning is crystalline GeO2 and finally turns to the amorphous germanium at the following cycles.
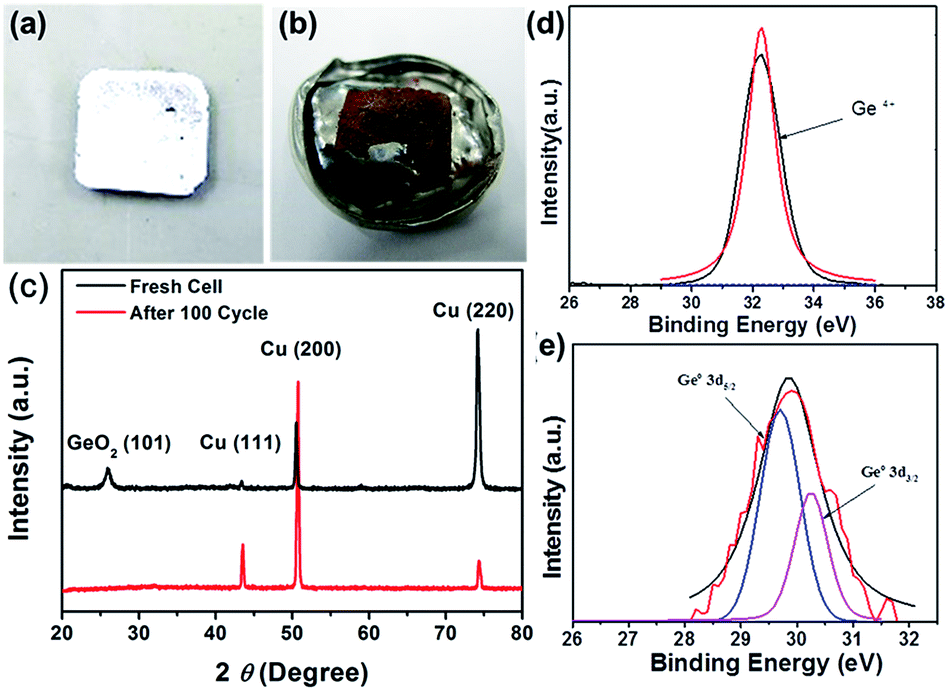 | ||
| Fig. 6 (a, b) Photographs of GeO2 anode color before/after 100th cycling intuitively (scale bar = 1 cm). (c) XRD pattern, (d, e) XPS analysis of GeO2 anode before/after 100th cycling. | ||
Volumetric capacity of active material is another essential consideration to evaluate the feasibility of the material applied to industrial development.59–61 According to the characteristics of GeO2 anode including gram-scale and great electrochemical properties analyzed in above experiments, the volumetric capacity of the GeO2 is around 660 mA h cm−3 on the basis of compress density 0.6 g cm−3 (active material weight, electrode area, and the thickness of the GeO2 electrode are 0.5 mg, 0.95 cm2, and ∼10 μm, respectively). This performance is higher than commercialized graphite anodes (370–500 mA h cm−3).62–64
Coin type and pouch type full cell were fabricated combined with cathode ternary material Li(NiCoMn)O2 to evaluate the feasibility of GeO2 nanoparticles toward its application. Fig. 7(a) show the cycle performance of GeO2 at rate of 0.1C in CR2032 coin type cells exhibited discharge capacities 1300 mA h g−1 in the first cycle between 2.5 V and 4.2 V and have a capacity of 950 mA h g−1 after 100 cycle. The subsequent slightly capacity fade is due to the unbalanced capacity ratio of cathode/anode and highly irreversible capacity of anodes. GeO2-based coin type full cell also perform well at the rate of 1C with reversible capacity of 600 mA h g−1. As for the pouch cell, the size of electrodes used in assembly is approximately 8 cm2 × 5 cm2, and the anode size is bigger to prevent Li-dendrite inside the cell. The mass loading of cathode and anode is the same as which is in CR2032 coin type cell. The total capacity and specific capacity are shown as Fig. 7(d). The capacity offered by the single pouch type battery at the rate 0.1C is 150 mA h (1878 mA h g−1) in the first cycle, and the subsequent cycles show reversible capacity of approximately 70 mA h (800 mA h g−1). To the best of our knowledge, it is the first time to incorporate the GeO2 nanoparticles into the pouch-type cell fabrication process. A single battery is shown to power LED array over 120 bulbs which correspond to driving current 660 mA, a discharge rate 3C–4C, (Fig. 7(e)).
 | ||
| Fig. 7 (a) Cycle performance of the Li(NiCoMn)O2/GeO2 CR2032 coin type full cell at a 0.1C rate between 2.5 V and 4.2 V. (b) Cycle performance of the Li(NiCoMn)O2/GeO2 CR2032 coin type full cell at a 1C rate between 2.5 V and 4.2 V. (c) Photography of Li(NiCoMn)O2/GeO2 pouch type full cell. (d) Cycle performance (total capacity) of the Li(NiCoMn)O2/GeO2 pouch type full cell at a 0.1C rate between 2.5 V and 4.2 V. (e) Demonstration of pouch-type full cell to power LEDs array with a maximum current of 650 mA. | ||
Conclusions
Nearly 100% yield of GeO2 nanoparticles was prepared by a green synthesis method in reverse micelle system at room temperature and exhibits superior electrochemical performance. As Li-ion batteries anode without any treatment, GeO2 nanoparticles developed in this work exhibit excellent gravimetric capacity (1050 mA h g−1 at 0.1C rate), and a good rate capability over tens of cycles, and perform well in volumetric capacity (660 mA h cm−3). Furthermore, coin type and pouch type full cells are successfully assembled with ternary cathode Li(NiCoMn)O2 to power a wide range of electronic application devices. The utilization of GeO2 as LIB anodes still have a challenging issue about low first coulombic efficiency induced by the irreversible process, which consume excess lithium ions to form Li2O. In the future, we believe that strategies to address the limitation of coulombic efficiency, combined with the scalable nanoparticle synthesis, high electrochemical performance in gravimetric and volumetric capacity, makes GeO2 a promising opportunity for the forthcoming low-cost and high energy density lithium-ion batteries.Acknowledgements
We acknowledge the financial support by the Ministry of Science and Technology through the grants of NSC 102-2221-E-007-023-MY3, MOST 103-2221-E-007-089-MY3, MOST 103-2622-E-007-025, and MOST 102-2633-M-007-002.Notes and references
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 CAS.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- M. Osiak, H. Geaney, E. Armstrong and C. O'Dwyer, J. Mater. Chem. A, 2014, 2, 9433–9460 CAS.
- F. W. Yuan, H. J. Yang and H. Y. Tuan, ACS Nano, 2012, 6, 9932–9942 CrossRef CAS PubMed.
- P. R. Abel, K. C. Klavetter, K. Jarvis, A. Heller and C. B. Mullins, J. Mater. Chem. A, 2014, 2, 19011–19018 CAS.
- J. G. Ren, Q. H. Wu, H. Tang, G. Hong, W. J. Zhang and S. T. Lee, J. Mater. Chem. A, 2013, 1, 1821–1826 CAS.
- D. Li, C. Q. Feng, H. K. Liu and Z. P. Guo, J. Mater. Chem. A, 2015, 3, 978–981 CAS.
- T. Kennedy, E. Mullane, H. Geaney, M. Osiak, C. O'Dwyer and K. M. Ryan, Nano Lett., 2014, 14, 716–723 CrossRef CAS PubMed.
- F. W. Yuan and H. Y. Tuan, Chem. Mater., 2014, 26, 2172–2179 CrossRef CAS.
- C. N. R. Rao and A. K. Cheetham, J. Mater. Chem., 2001, 11, 2887–2894 RSC.
- H. B. Wu, J. S. Chen, H. H. Hng and X. W. Lou, Nanoscale, 2012, 4, 2526–2542 RSC.
- M. Z. Ge, C. Y. Cao, J. Y. Huang, S. H. Li, Z. Chen, K. Q. Zhang, S. S. Al-Deyabd and Y. K. Lai, J. Mater. Chem. A, 2016, 4, 6772–6801 CAS.
- P. Roy and S. K. Srivastava, J. Mater. Chem. A, 2015, 3, 2454–2484 CAS.
- X. M. Lu, D. D. Fanfair, K. P. Johnston and B. A. Korgel, J. Am. Chem. Soc., 2005, 127, 15718–15719 CrossRef CAS PubMed.
- J. M. Buriak, Chem. Rev., 2002, 102, 1271–1308 CrossRef CAS PubMed.
- S. V. Patwardhan and S. J. Clarson, Polymer, 2005, 46, 4474–4479 CrossRef CAS.
- A. R. Phani, D. Di Claudio, M. Passacantando and S. Santucci, J. Non-Cryst. Solids, 2007, 353, 692–696 CrossRef CAS.
- H. Z. Li, L. Y. Yang, J. Liu, S. T. Li, L. B. Fang, Y. K. Lu, H. R. Yang, S. L. Liu and M. Lei, J. Power Sources, 2016, 324, 780–787 CrossRef CAS.
- J. Liu, P. J. Lu, S. Liang, J. Liu, W. Wang, M. Lei, S. Tang and Q. Yang, Nano Energy, 2015, 12, 709–724 CrossRef CAS.
- J. Liu, S. S. Tang, Y. K. Lu, G. M. Cai, S. Q. Liang, W. J. Wang and X. L. Chen, Energy Environ. Sci., 2013, 6, 2691–2697 CAS.
- P. J. Lu, M. Lei and J. Liu, CrystEngComm, 2014, 16, 6745–6755 RSC.
- L. Mei, M. L. Mao, S. L. Chou, H. K. Liu, S. X. Dou, D. H. L. Ng and J. M. Ma, J. Mater. Chem. A, 2015, 3, 21699–21705 CAS.
- H. Y. Qiu, L. X. Zeng, T. B. Lan, X. K. Ding and M. D. Wei, J. Mater. Chem. A, 2015, 3, 1619–1623 CAS.
- J. S. Pena, I. Sandu, O. Joubert, F. S. Pascual, C. O. Arean and T. Brousse, Electrochem. Solid-State Lett., 2004, 7, A278–A281 CrossRef CAS.
- L. X. Zeng, X. X. Huang, X. Chen, C. Zheng, Q. R. Qian, Q. H. Chen and M. D. Wei, ACS Appl. Mater. Interfaces, 2016, 8, 232–239 CAS.
- A. Jahel, A. Darwiche, C. M. Ghimbeu, C. Vix-Guterl and L. Monconduit, J. Power Sources, 2014, 269, 755–759 CrossRef CAS.
- S. Yoon, S. H. Jung, K. N. Jung, S. G. Woo, W. Cho, Y. N. Jo and K. Y. Cho, Electrochim. Acta, 2016, 188, 120–125 CrossRef CAS.
- D. T. Ngo, H. T. T. Le, R. S. Kalubarme, J. Y. Lee, C. N. Park and C. J. Park, J. Mater. Chem. A, 2015, 3, 21722–21732 CAS.
- J. Hwang, C. Jo, M. G. Kim, J. Chun, E. Lim, S. Kim, S. Jeong, Y. Kim and J. Lee, ACS Nano, 2015, 9, 5299–5309 CrossRef CAS PubMed.
- Y. Chen, C. L. Yan and O. G. Schmidt, Adv. Energy Mater., 2013, 3, 1269–1274 CrossRef CAS.
- Y. Son, M. Park, Y. Son, J. S. Lee, J. H. Jang, Y. Kim and J. Cho, Nano Lett., 2014, 14, 1005–1010 CrossRef CAS PubMed.
- Y. M. Lin, K. C. Klavetter, A. Heller and C. B. Mullins, J. Phys. Chem. Lett., 2013, 4, 999–1004 CrossRef CAS PubMed.
- X. L. Wang, W. Q. Han, H. Y. Chen, J. M. Bai, T. A. Tyson, X. Q. Yu, X. J. Wang and X. Q. Yang, J. Am. Chem. Soc., 2011, 133, 20692–20695 CrossRef CAS PubMed.
- S. L. Shinde and K. K. Nanda, CrystEngComm, 2013, 15, 1043–1046 RSC.
- M. Gunji, S. V. Thombare, S. Hu and P. C. McIntyre, Nanotechnology, 2012, 23, 385603 CrossRef CAS PubMed.
- Y. J. Zhang, J. Zhu, Q. Zhang, Y. J. Yan, N. L. Wang and X. Z. Zhang, Chem. Phys. Lett., 2000, 317, 504–509 CrossRef CAS.
- Q. Y. Lu, F. Gao, Y. Q. Li, Y. M. Zhou and D. Y. Zhao, Microporous Mesoporous Mater., 2002, 56, 219–225 CrossRef CAS.
- X. D. Zou, T. Conradsson, M. Klingstedt, M. S. Dadachov and M. O'Keeffe, Nature, 2005, 437, 716–719 CrossRef CAS PubMed.
- Z. Jiang, T. Xie, G. Z. Wang, X. Y. Yuan, C. H. Ye, W. P. Cai, G. W. Meng, G. H. Li and L. D. Zhang, Mater. Lett., 2005, 59, 416–419 CrossRef CAS.
- Y. W. Chiu and M. H. Huang, J. Phys. Chem. C, 2009, 113, 6056–6060 CAS.
- N. Liu, H. Wu, M. T. McDowell, Y. Yao, C. M. Wang and Y. Cui, Nano Lett., 2012, 12, 3315–3321 CrossRef CAS PubMed.
- Y. R. Wang, W. Zhou, L. P. Zhang, G. S. Song and S. Q. Cheng, RSC Adv., 2015, 5, 63012–63016 RSC.
- N. Yan, F. Wang, H. Zhong, Y. Li, Y. Wang, L. Hu and Q. W. Chen, Sci. Rep., 2013, 3, 1568 Search PubMed.
- K. H. Seng, M. H. Park, Z. P. Guo, H. K. Liu and J. Cho, Nano Lett., 2013, 13, 1230–1236 CrossRef CAS PubMed.
- C. H. Kim, Y. S. Jung, K. T. Lee, J. H. Ku and S. M. Oh, Electrochim. Acta, 2009, 54, 4371–4377 CrossRef CAS.
- Y. G. Zhu, Y. Wang, Z. J. Han, Y. Shi, J. I. Wong, Z. X. Huang, K. Ostrikov and H. Y. Yang, Nanoscale, 2014, 6, 15020–15028 RSC.
- D. T. Ngo, R. S. Kalubarme, H. T. T. Le, C. N. Park and C. J. Park, Nanoscale, 2015, 7, 2552–2560 RSC.
- D. T. Ngo, R. S. Kalubarme, M. G. Chourashiya, C. N. Park and C. J. Park, Electrochim. Acta, 2014, 116, 203–209 CrossRef CAS.
- H. P. Jia, R. Kloepsch, X. He, J. P. Badillo, M. Winter and T. Placke, J. Mater. Chem. A, 2014, 2, 17545–17550 CAS.
- Y. G. Li, B. Tan and Y. Y. Wu, Nano Lett., 2008, 8, 265–270 CrossRef CAS PubMed.
- J. R. Dahn, T. Zheng, Y. H. Liu and J. S. Xue, Science, 1995, 270, 590–593 CAS.
- A. M. Chockla, K. C. Klavetter, C. B. Mullins and B. A. Korgel, ACS Appl. Mater. Interfaces, 2012, 4, 4658–4664 CAS.
- S. S. Zhang, J. Power Sources, 2006, 162, 1379–1394 CrossRef CAS.
- Y. M. Lin, K. C. Klavetter, P. R. Abel, N. C. Davy, J. L. Snider, A. Heller and C. B. Mullins, Chem. Commun., 2012, 48, 7268–7270 RSC.
- M. Itagaki, K. Honda, Y. Hoshi and I. Shitanda, J. Electroanal. Chem., 2015, 737, 78–84 CrossRef CAS.
- Y. W. Li, J. H. Yao, E. Uchaker, J. W. Yang, Y. X. Huang, M. Zhang and G. Z. Cao, Adv. Energy Mater., 2013, 3, 1171–1175 CrossRef CAS.
- B. Luo, Y. Fang, B. Wang, J. S. Zhou, H. H. Song and L. J. Zhi, Energy Environ. Sci., 2012, 5, 5226–5230 CAS.
- X. H. Wang, L. N. Sun, R. A. Susantyoko, Y. Fan and Q. Zhang, Nano Energy, 2014, 8, 71–77 CrossRef CAS.
- H. L. Cao, X. F. Zhou, W. Deng and Z. P. Liu, J. Mater. Chem. A, 2016, 4, 6021–6028 CAS.
- K. C. Klavetter, J. P. de Souza, A. Heller and C. B. Mullins, J. Mater. Chem. A, 2015, 3, 5829–5834 CAS.
- S. Jeong, J. P. Lee, M. Ko, G. Kim, S. Park and J. Cho, Nano Lett., 2013, 13, 3403–3407 CrossRef CAS PubMed.
- B. Wang, X. L. Li, T. F. Qiu, B. Luo, J. Ning, J. Li, X. F. Zhang, M. H. Liang and L. J. Zhi, Nano Lett., 2013, 13, 5578–5584 CrossRef CAS PubMed.
- X. P. Wang, L. X. Lv, Z. H. Cheng, J. A. Gao, L. Y. Dong, C. G. Hu and L. T. Qu, Adv. Energy Mater., 2016, 6, 1502100 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Cycling life curve for GeO2 nanoparticles at different electrolyte systems at 0.1C and 1C. See DOI: 10.1039/c6ra20171g |
| This journal is © The Royal Society of Chemistry 2016 |