DOI:
10.1039/C6RA05478A
(Paper)
RSC Adv., 2016,
6, 98620-98631
Macrocyclization of N,N′-propylenebis(3-formyl-5-tert-butylsalicylaldimine): a ratiometric fluorescence chemodosimeter for ZnII†
Received
1st March 2016
, Accepted 8th October 2016
First published on 12th October 2016
Abstract
Addition of 1,3-propane diamine to 2,6-diformyl-4-tert-butyl phenol in ethanol produces a site-selective imination product N,N′-propylenebis(3-formyl-5-tert-butylsalicylaldimine), an acyclic side-off compartmental ligand (H2L). In the presence of zinc nitrate the ligand goes on hydrolysis in 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 water–acetonitrile medium and forms a partially hydrolyzed ligand (H2L′) which slowly metallates to generate a macrocyclic dinuclear zinc(II) complex (1), as characterized by single crystal X-ray analyses. The formation of H2L′ is believed to occur through the cleavage of an imine bond of the acyclic compartmental ligand (H2L) in the presence of zinc nitrate which acts as a Lewis acid. The formation of H2L′ has been monitored by means of 1H NMR and further confirmed by HRMS spectroscopic studies. The interactions of H2L with nickel(II) and copper(II) nitrate produce dinuclear complexes 2 and 3 (reported in Inorg. Chem. Commun. 2012, 15, 266–268) respectively, which are formed with unchanged ligand. Various spectroscopic techniques have been used to further characterize the complexes. H2L hardly exhibits yellowish green fluorescence emission at 523 nm when excited at 437 nm in 1:1 water–acetonitrile. Upon addition of Zn2+, a new fluorescence emission band at 481 nm appears, the intensity of which slowly enhances. Thus, the ligand H2L is a ratiometric fluorescence chemodosimeter for the selective detection of Zn2+ ions. On addition of CaII, MgII, NaI and KI in the same concentrations as that of Zn2+, the emission band at 523 nm is slightly enhanced, whereas the addition of paramagnetic metal cations like CuII, FeII, NiII, CoII, and MnII resulted in quenching of fluorescence. The quenching effect is also observed in the presence of CdII, a d10 metal cation exhibiting similar coordination properties to ZnII. The ZnII ion selectivity has also been studied in the presence of other biologically relevant metal ions in 50:50 water–acetonitrile.
:50 water–acetonitrile medium and forms a partially hydrolyzed ligand (H2L′) which slowly metallates to generate a macrocyclic dinuclear zinc(II) complex (1), as characterized by single crystal X-ray analyses. The formation of H2L′ is believed to occur through the cleavage of an imine bond of the acyclic compartmental ligand (H2L) in the presence of zinc nitrate which acts as a Lewis acid. The formation of H2L′ has been monitored by means of 1H NMR and further confirmed by HRMS spectroscopic studies. The interactions of H2L with nickel(II) and copper(II) nitrate produce dinuclear complexes 2 and 3 (reported in Inorg. Chem. Commun. 2012, 15, 266–268) respectively, which are formed with unchanged ligand. Various spectroscopic techniques have been used to further characterize the complexes. H2L hardly exhibits yellowish green fluorescence emission at 523 nm when excited at 437 nm in 1:1 water–acetonitrile. Upon addition of Zn2+, a new fluorescence emission band at 481 nm appears, the intensity of which slowly enhances. Thus, the ligand H2L is a ratiometric fluorescence chemodosimeter for the selective detection of Zn2+ ions. On addition of CaII, MgII, NaI and KI in the same concentrations as that of Zn2+, the emission band at 523 nm is slightly enhanced, whereas the addition of paramagnetic metal cations like CuII, FeII, NiII, CoII, and MnII resulted in quenching of fluorescence. The quenching effect is also observed in the presence of CdII, a d10 metal cation exhibiting similar coordination properties to ZnII. The ZnII ion selectivity has also been studied in the presence of other biologically relevant metal ions in 50:50 water–acetonitrile.
Introduction
The design and synthesis of metal ion sensing fluorescent chemodosimeters have become engrossing areas of research because of their greater advantages over chemosensors.1–4 Considerable efforts have been devoted in the past few years to developing metal sensing chemodosimeters after the first introduction by Chae and Czarnik in 1992.5–12 The principal idea of this system is to take advantage of the selective reactivity that a certain cation may display over the others in an irreversible way. As the second most abundant transition metal in the human body, zinc plays a crucial role in various biological processes such as gene transcription, regulation of metalloenzymes and neural signal transmission.13 Environmentally, free zinc ions present in solution are highly toxic to bacteria, plants, invertebrates and even vertebrate fish.14 Hence, the development of Zn2+ ion sensing fluorescent probes is a promising field. Few reports of chemodosimetric fluorescent probes for zinc metal have appeared in the literature, most of which, however, are based on enhancement in fluorescence intensity.15,16 Although turn-on probes have high sensitivity due to the lack of background signal, a major limitation of intensity-based probes is that variations in the sample environment may influence the measurements of fluorescence intensity. In principle, this problem can be alleviated by using ratiometric fluorescent probes. Numerous ratiometric fluorescence chemodosimeters are available in literature,17,18 but very limited for ZnII.19 The ratiometric fluorescence probes allow the measurement of fluorescence intensities at two different wavelengths which should provide a built-in correction for environmental effects.17 Ratiometric measurements are also independent of the probe concentration, path length, or spectral sensitivity of the instrument.18
Because of the metal–metal distance found in their metal building blocks, 2,6-diformyl 4-substituted phenol based Schiff base compartmental ligands have widely been used to design mimics of various metalloenzyme in biological systems.20–25 In spite of possessing high fluorescence property Schiff base compartmental ligands have not been considered sufficiently as fluorescent probes for cations.26–28 Considering the above, we judiciously designed an acyclic phenol-based Schiff base compartmental ligand, N,N′-propylene-bis(3-formyl-5-tert-butyl-salicylaldimine) with formyl group in one arm (see Scheme 1) as the reaction unit with Zn2+. It is needless to say that compounds containing acyclic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds are highly susceptible towards hydrolysis and tend to form a stable cyclic compound in presence of ZnCl2 like Lewis acid in organic-aqua medium.
N bonds are highly susceptible towards hydrolysis and tend to form a stable cyclic compound in presence of ZnCl2 like Lewis acid in organic-aqua medium.
 |
| | Scheme 1 Acyclic compartmental ligand (H2L). | |
Results and discussions
Syntheses of ligand and complexes
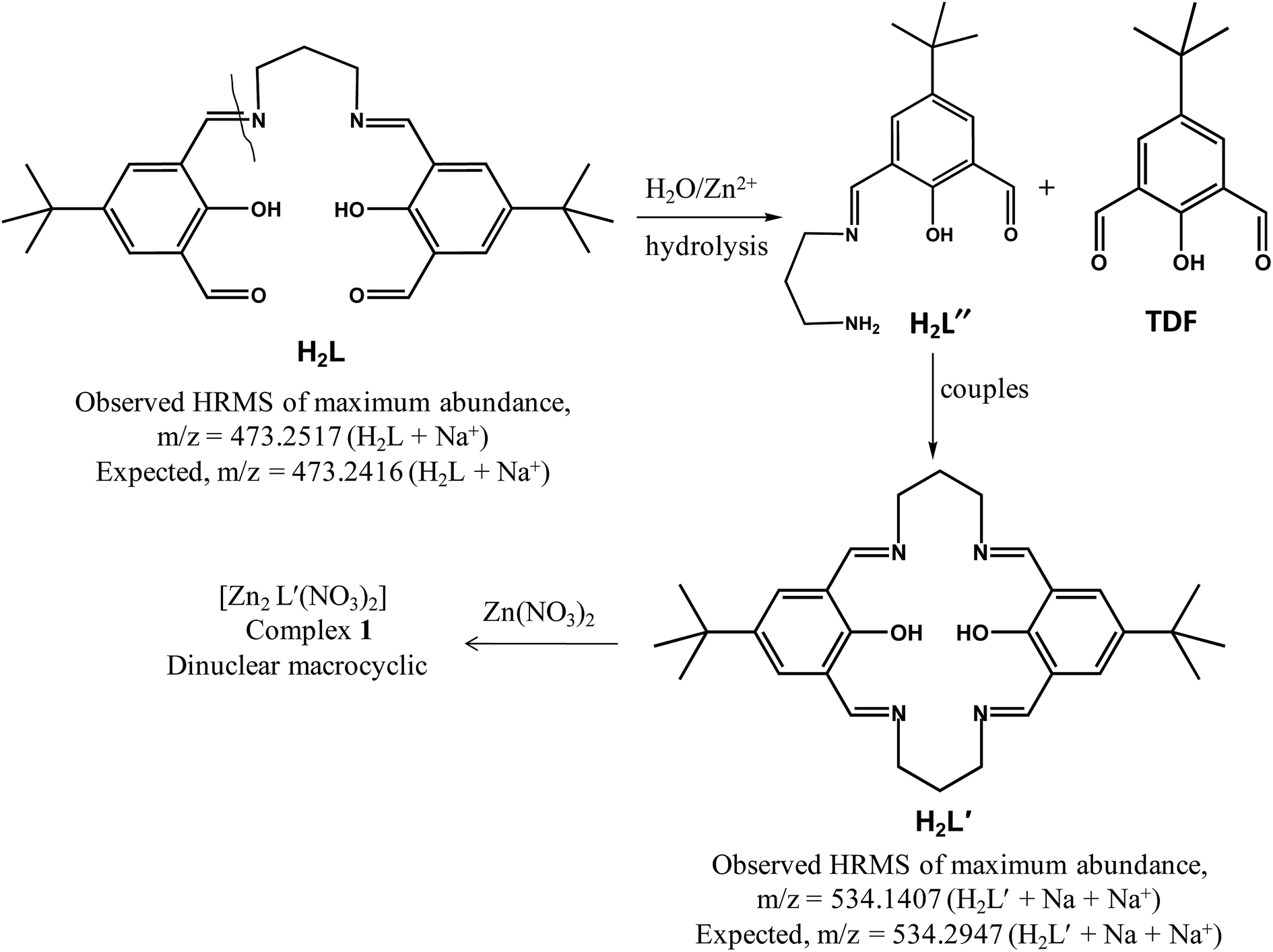
The reactions of side-off ligand N,N′-propylenebis(3-formyl-5-tert-butylsalicylaldimine) (H2L) with different metal(II) nitrates have been systematically investigated and only in three instances (in case of NiII, CuII and ZnII) we are able to find out crystal structures as summarized in Scheme 2. In presence of zinc nitrate, macrocyclic ligand (H2L′) is formed in 50:50 water–acetonitrile medium, which in turn metallates to generate a macrocyclic dinuclear zinc(II) complex (1). The formation of H2L′ is believed to occur through the hydrolysis of one imine (CN) arm of the acyclic compartmental ligand (H2L) in presence of zinc nitrate which acts as a Lewis acid. Despite several ZnII/H2L stoichiometric ratios (from 1:1 to 1:3) being used, in each instance dinuclear macrocyclic zinc(II) complex is found to be formed. On the contrary, interaction of copper(II) nitrate and nickel(II) nitrate with H2L produce dinuclear side-off complexes where no hydrolysis had taken place even at higher MII/H2L stoichiometric ratios (upto 1:3).
 |
| | Scheme 2 Synthetic routes of complexes 1–3. | |
Characterization of ligand and complexes
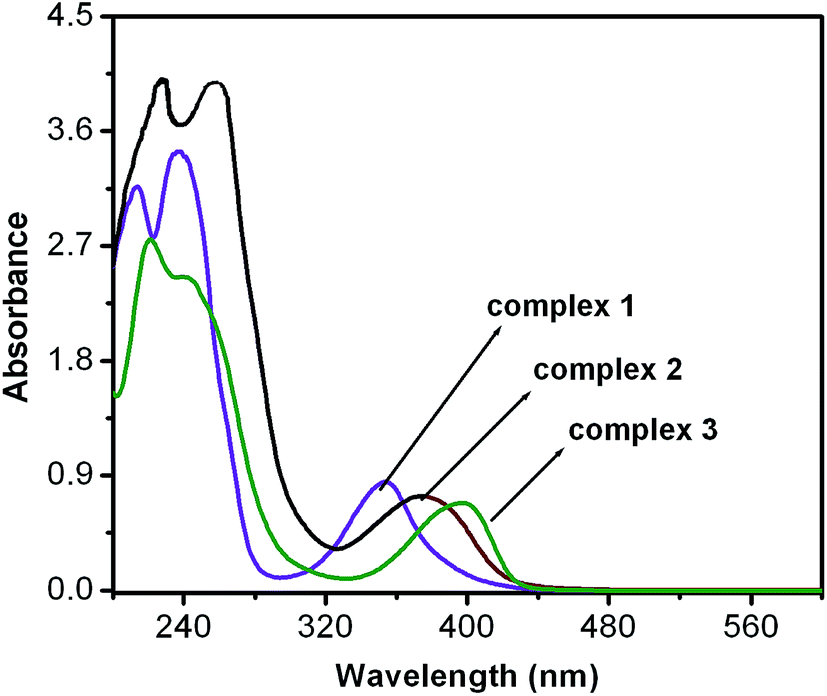
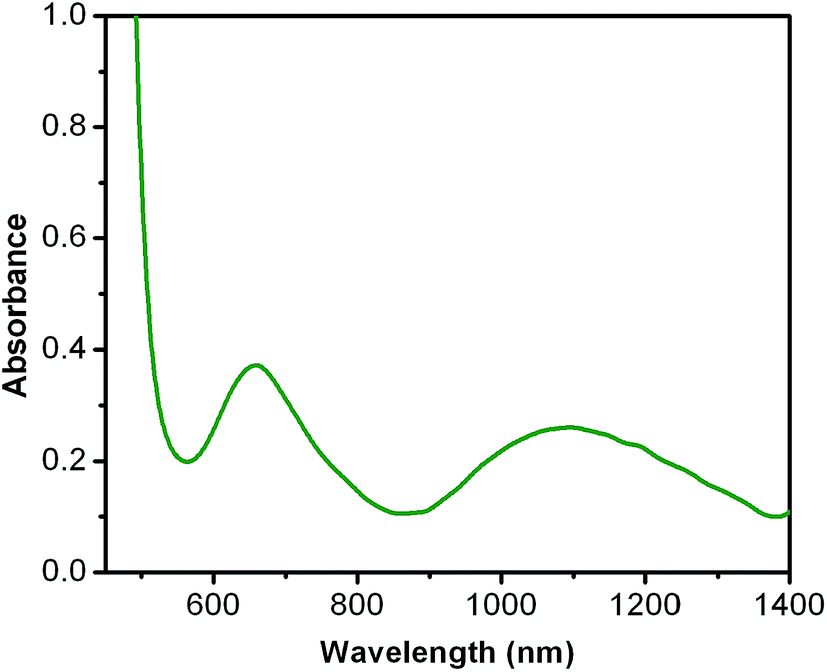
Electronic spectra. Electronic spectra of all the complexes are shown in Fig. 1–3. Complex 1 shows three types of band in acetonitrile. Two very intense band at 237 nm (ε = 34283 L mol−1 cm−1) and 214 nm (ε = 31419 L mol−1 cm−1) are dominated by intra ligand charge-transfer transition (π–π* transition) and a shoulder at 353 nm (ε = 8454 L mol−1 cm−1) may be due to PhO− to ZnII ligand-to-metal charge-transfer transition.29,30 In complex 2, three characteristics absorption bands appeared in acetonitrile solution (Fig. 2). A broad absorption band at 748 nm (ε = 40.3 L mol−1 cm−1) has been appeared, related to the ligand field spectra induced by the H2L ligand. The other two bands, one at 245 nm (ε = 42098 L mol−1 cm−1) is dominated by intra ligand charge-transfer transition and another shoulder at 374 nm (ε = 7504 L mol−1 cm−1) is due to PhO− to CuII ligand-to-metal charge-transfer transition. Complex 3 in acetonitrile solution shows multiple bands in the 200–1400 nm region (Fig. 3). Two broad absorption bands are observed at 658 nm (ε = 37.5 L mol−1 cm−1) and 1094 nm (ε = 26.2 L mol−1 cm−1). The former (near 640 nm) is due to the ligand field transitions induced by the ligand (H2L) and other is responsible for the transition of 3A2g → 3T2g(F), respectively. Another two intense absorption bands at 241 nm (ε = 24406 L mol−1 cm−1) and 221 nm (ε = 27431 L mol−1 cm−1) are dominated by intra ligand charge-transfer transition. Spectrum of complex 3 also shows a shoulder at 395 nm (ε = 6862 L mol−1 cm−1) which is due to PhO− to NiII ligand-to-metal charge-transfer transition.
 |
| | Fig. 1 UV-Vis spectra of 10−4 M solution of complexes 1–3 in acetonitrile. | |
 |
| | Fig. 2 UV-Vis spectra of 10−2 M solution of complex 2 in acetonitrile. | |
 |
| | Fig. 3 UV-Vis spectra of 10−2 M solution of complex 3 in acetonitrile. | |
FTIR spectra. Side off Schiff base ligand (H2L) has three characteristic IR bands at 1682, 1633 and 1599 cm−1 are assigned to CO, CN and skeletal vibrations (Fig. S1, ESI†), respectively similar to our earlier report.31 IR spectra of complex 1 showed two sharp bands, one at 1597.4 cm−1 and another at 1496.9 cm−1 due to characteristic stretching of νCN of macrocyclic Schiff base ligand (H2L′) and skeletal benzene ring respectively (Fig. S2, ESI†). Absence of peak around 1682 cm−1 in complex 1 suggests the conversion of free formyl group into imine bond during complexation. A sharp band centered at 1338.9 cm−1 is assigned to unsymmetrical stretching frequency of nitrate anion coordinated to ZnII by monodentate fashion. In complex 2, the characteristic bands of νCN and νCHO of ligand (H2L) are detected at 1625.6 and 1638 cm−1 respectively (Fig. S3, ESI†). The skeletal frequency of benzene ring moiety is observed at 1546.8 cm−1. A sharp band at 1463 cm−1 due to symmetric stretching frequency of bidentate bridging nitrate anion was also observed. In complex 3, the characteristic bands of νCN of Schiff base ligand (H2L) (Fig. S4, ESI†) and the skeletal benzene ring moiety are appeared at 1645.5 cm−1 and at 1544.5 cm−1 respectively. In 1981 Gagne et al.32 prepared heterodinuclear complex (MAMBL2+) from a ligand, very similar to that we have used here in this work, and they confirmed the formation of MAMBL2+ by IR and EPR spectral studies. They observe bands at ∼1645, ∼1600 and 1550 cm−1, corresponding to νCO, νCN and skeletal vibrations respectively. Unfortunately, we fail to observe the characteristic band of νCO(aldehyde) of the ligand (H2L) probably because these two bands are merged together. A broad band centered at 1362.8 cm−1 was assign to unsymmetrical stretching frequency of nitrate anion coordinated to NiII by monodentate fashion.
Complexation studies
Apparent metal(II) binding affinity by using Job's method. The binding affinities of the ligand towards various metals are directly obtained from emission intensity versus mol-ratio plots with metal(II) concentrations ranging from 0 to 200 μm. Job's analysis33 showed 1:1 binding of metal to ligand for ZnII (Fig. 4) whereas 1:2 binding for 1st row transition metals. Only the distinct behavior of ZnII to N,N′-propylenebis(3-formyl-5-tert-butylsalicylaldimine) has been studied in details by 1H NMR and ESI-MASS analysis in 1:1 water–acetonitrile.
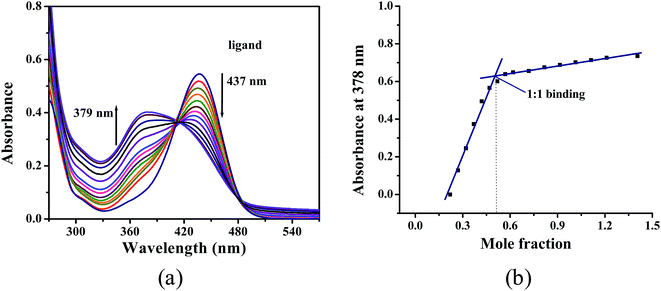 |
| | Fig. 4 (a) Spectrophotometric titration of ligand, H2L (1 × 10−4 M) in presence of increasing concentrations of zinc nitrate (0–2 × 10−4 M); (b) Job's plot (absorbance vs. mole fraction of Zn2+) in 50:50 acetonitrile. | |
Complexation study by 1H NMR spectra. In order to confirm the ZnII catalyzed macrocyclic ligand (H2L′) formation from the mother ligand (H2L), 1H NMR titration is performed in CD3CN solvent (Fig. 5a). The observed signals for the mother ligand (H2L) are at 10.3 (Ha), 8.32 (Hb), 7.68 (Hc), 7.4 (Hd), 3.6 (Hf), 1.98 (He) and 1.15 (Hg) ppm. After addition of zinc nitrate the reaction mixture is equilibrated 30 min and the spectrum is recorded. The observed signals are found to be shifted than before and positioned at 10.19 (Ha), 7.8 (Hb), 8.2 (Hc), 7.45 (Hd), 3.98 (Hf), 2.18 (He) and 1.2 (Hg) ppm. At sudden look these two spectral patterns seem to be similar. If we analyze the peaks at 1.15 (Hg) (Fig. 5a) and 1.2 (Hg) ppm (Fig. 5b) carefully it would be found that there are two types of tert-butyl protons in the spectrum obtained from ligand + zinc nitrate solution. The integration of these two individual peaks does not match well with the other peaks in the corresponding spectra. Yet, these spectra tell worth about the presence of both macrocyclic ligand (H2L′) and 2,6-diformyl 4-tert butyl phenol formed in the solution due to hydrolysis of mother ligand (H2L). NMR spectrum with pure Zn complex was also taken in same solvent (Fig. S5, ESI†) and the spectrum well correlate with Fig. 5b, suggesting that there are the same compounds in both the cases. Comparison of 13C NMR of the mixture and pure ligand clearly tells a interaction is playing between ligand and zinc ion (Fig. S6, ESI†).
 |
| | Fig. 5 1H NMR spectra of (a) ligand (H2L) only and (b) ligand (H2L) + Zn(NO3)2 in acetonitrile medium recorded after 30 min of mixing. | |
Complexation study by ESI-MS mass spectra. For further confirmation of the formation of macrocyclic ligand (H2L′) from the mother ligand (H2L), ESI-MS mass spectral study was employed. ESI-MS positive spectrum of only H2L in 50:50 water–acetonitrile medium shows one major peak at m/z = 451.2531 with line-to-line separation of 1.0 can be assigned as the [H2L + H+] (Fig. S7, ESI†). In order to get an insight into the possible intermediates of this transformation, the ESI-MS positive spectrum of a 1:1 mixture of the ligand H2L and zinc nitrate in 50:50 water–acetonitrile was recorded after 5 minutes of mixing, and the spectra is depicted in Fig. 6. A prominent peak with maximum abundance observed at m/z = 534.1476 with line-to-line separation of 1.0 corresponds to a unipositive species [H2L′ + Na + Na+] (Fig. 6). The mass spectral study clearly indicates that the macrocyclic ligand H2L′ was formed in the reaction between ligand (H2L) and zinc nitrate. Another two important peaks at m/z = 207.0846 and 245.0883 can be assigned as the [TDF + H+] and [TDF + K+], respectively. One interesting peak at m/z = 263.1574 can be attributed as monoprotonated species of 1:1 condensed product of 1,3-propane diammine and 2,6-diformyl-4-tert-butylphenol, namely [H2L′′ + H+]. After 20 minutes the mixture generates a major peak at m/z = 680, indicating the formation of complex 1 (Fig. S8, ESI†). The ESI-MS spectrum of zinc complex (1) in 50:50 water–acetonitrile medium shows two major peaks at m/z = 309.1555 and 676.1589 can be attributed as [H2L′′ + Na + Na+] and the molecular ion [L′Zn2NO3+], respectively (Fig. S9, ESI†). The experimentally observed and the simulated spectral patterns are in excellent agreement with each other, indicating right assignment of the intermediate species.
 |
| | Fig. 6 ESI-MS spectra of ligand and zinc nitrate mixture in 50:50 water–acetonitrile after 5 minutes of mixing. | |
From the above experiments, it may be concluded that the addition of zinc nitrate to a solution of ligand (H2L) in 50:50 water–acetonitrile undergoes hydrolysis into monocondensed ligand H2L′′ and TDF. H2L′′ further endure coupling to generate macrocyclic ligand H2L′. Reaction between macrocyclic ligand H2L′ with zinc nitrate construct a dinuclear macro-cyclic zinc(II) complex [Zn2(L′)(NO3)2] (characterized by single crystal X-ray analysis as well as ESI-MS spectroscopy). The whole phenomenon may be called as zinc ion induced dissociation–aggregation process. Possible reaction mechanism for the macrocyclic zinc complex (1) is depicted below in Scheme 3.
 |
| | Scheme 3 Proposed mechanism for the formation of dinuclear macrocyclic complex 1. | |
Structural description of complexes 1 and 2
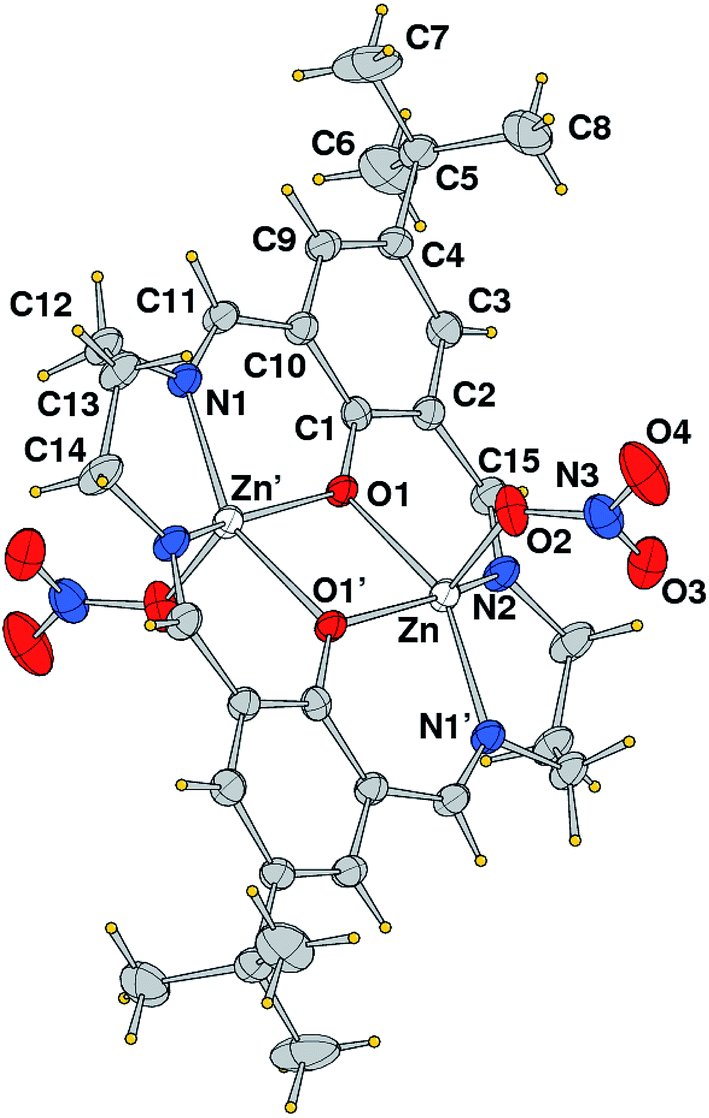
The single crystal diffraction analysis reveals that complex 1 crystallizes in the monoclinic space group P21/n. The crystal structure analysis shows that it is a centrosymmetric dinuclear zinc(II) complex (Fig. 7) with macrocyclic ligand having a N4O2 donor set, thus a ligand backbone different from that of the mother one. The six donors of Schiff base occupy the basal positions of the distorted square pyramidal coordination geometry of the zinc ions. At the axial position resides an oxygen atom from nitrate anion connected in monodentate fashion. The complex is located on a center of symmetry, thus both zinc atoms have same coordination environment and identical bond angles and distances that are provided in Table 1. The Zn–O and Zn–N bond distances fall in the range from 2.062(3) to 2.074(2) Å, slightly longer than the axial one, of 2.026(3) Å. The metal–metal distance in the dinuclear core is of 3.2578(10) Å, and both metal ions are displaced by 0.463 Å from the respective basal mean plane towards nitrate oxygen atom. The Zn2O(phenoxo)2 coreplane is slightly tilted with respect to the mean plane through each benzene ring forming a dihedral angle of 23.25°.
 |
| | Fig. 7 Molecular structure (ORTEP drawing, 35% ellipsoid probability) of the centro-symmetric complex 1 with label scheme of the independent unit. | |
Table 1 Coordination bond lengths (Å) and angles (°) for complex 1
| Zn–O1 |
2.066(2) |
Zn–N1′ |
2.051(3) |
| Zn–O1′ |
2.074(2) |
Zn–N2 |
2.062(3) |
| Zn–O2 |
2.026(3) |
Zn–Zn′ |
3.2578(10) |
| O2–Zn–N1′ |
110.97(12) |
N2–Zn–O1 |
87.87(10) |
| O2–Zn–N2 |
111.39(12) |
O2–Zn–O1′ |
92.32(10) |
| N1′–Zn–N2 |
96.06(11) |
N1′–Zn–O1′ |
88.22(10) |
| O2–Zn–O1 |
93.23(11) |
N2–Zn–O1′ |
152.25(11) |
| N1′–Zn–O1 |
151.82(11) |
O1–Zn–O1′ |
76.18(9) |
Complex 2 crystallizes in the monoclinic space group C2. Crystal structure of the complex located on a crystallographic two-fold axis is shown in Fig. 8 and important bond lengths and angles are provided in Table 2. The ligand backbone of this complex is H2L, thus the unhydrolysed form of ligand observed in complex 1. The hexadentate Schiff base with N2O4 donor set coordinates two CuII ions that exhibit a distorted octahedral geometry, with axial positions occupied by oxygen atoms from nitrate anions coordinated in bridging fashion. The two Cu2–N(imine) bond distances are equal for symmetry (of 1.942(8) Å), where as the two pairs of Cu–O(phenoxo) bond distances are 1.933(5) and 1.962(5) Å for Cu1 and Cu2, respectively. The axial Cu1–O and Cu2–O bond distances are almost identical, of 2.483(6) and 2.492(7) Å, respectively and significantly longer of the equatorial ones. The intermetallic distance in the dinuclear core is 2.9707(9) Å. Taking into account the benzene at higher occupancy (atoms C1–6), the plane through the Cu2O2(phenoxo) core forms a dihedral angle of 13.24° with that through the benzene ring, so that the overall complex assumes a slight bowed shape. Crystal structure of reported NiII complex 3 is depicted in Fig. 9.
 |
| | Fig. 8 Molecular structure (ORTEP drawing, 35% ellipsoid probability) of complex 2 with label scheme of the independent unit. Metal atoms are located on a crystallographic two-fold axis that relates the two phenolate moieties. Of disorder group (see Experimental section) only atoms at higher occupancy are shown. | |
Table 2 Coordination bond lengths (Å) and angles (°) for complex 2
| Cu1–O1 |
1.933(6) |
Cu2–O1 |
1.962(6) |
| Cu1–O2 |
1.938(6) |
Cu2–N2 |
1.942(8) |
| Cu1–O3 |
2.483(6) |
Cu2–O4 |
2.492(7) |
| Cu1–Cu2 |
2.9707(9) |
|
|
| O1′–Cu1–O1 |
81.3(3) |
N2′–Cu2–N2 |
98.4(4) |
| O1–Cu1–O2 |
93.4(2) |
N2–Cu2–O1′ |
170.2(3) |
| O1–Cu1–O2′ |
172.4(3) |
N2–Cu2–O1 |
90.9(3) |
| O2′–Cu1–O2 |
92.4(4) |
O1′–Cu2–O1 |
79.9(3) |
| O1–Cu1–O3 |
83.11(17) |
O1–Cu2–O4 |
82.38(19) |
| O1–Cu1–O3′ |
84.01(17) |
O1–Cu2–O4′ |
82.25(19) |
| O2–Cu1–O3 |
90.0(3) |
O4–Cu2–N2 |
93.2(3) |
| O2–Cu1–O3′ |
101.8(3) |
O4–Cu2–O4′ |
159.9(2) |
| O3–Cu1–O3′ |
162.99(17) |
O4–Cu2–N2′ |
99.9(3) |
| Cu1–O1–Cu2 |
99.40(14) |
|
|
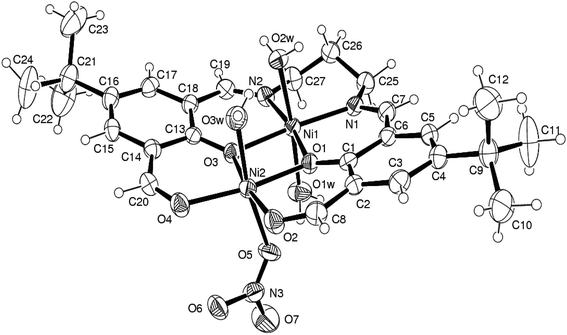 |
| | Fig. 9 ORTEP drawing (ellipsoids at 35% probability) of cationic form of complex 3 adapted from ref. 34. | |
Preparation of test solutions for fluorescence titration
The initial stock solution (1 × 10−3 M) of ligand H2L has been prepared in 50:50 water–acetonitrile medium. The solution is further diluted to 5 × 10−6 M and is used as a final stock for fluorescence titration. The final stock solutions of various cations (in the form of nitrate salt) are prepared at 5 × 10−4 M by dilution technique from an initial stock of 0.1 M each in 50:50 water–acetonitrile medium. In each individual titration experiment, 2 mL test solution contains 1 mL of stock ligand (5 × 10−6 M), 1 mL of 50:50 water–acetonitrile and 4–40 μL of corresponding cationic solution from 5 × 10−4 M stock. The resulting solution is shaken well and incubated for 10 min at room temperature before the titration experiment starts.
Electronic and emission spectra of ligand (H2L)
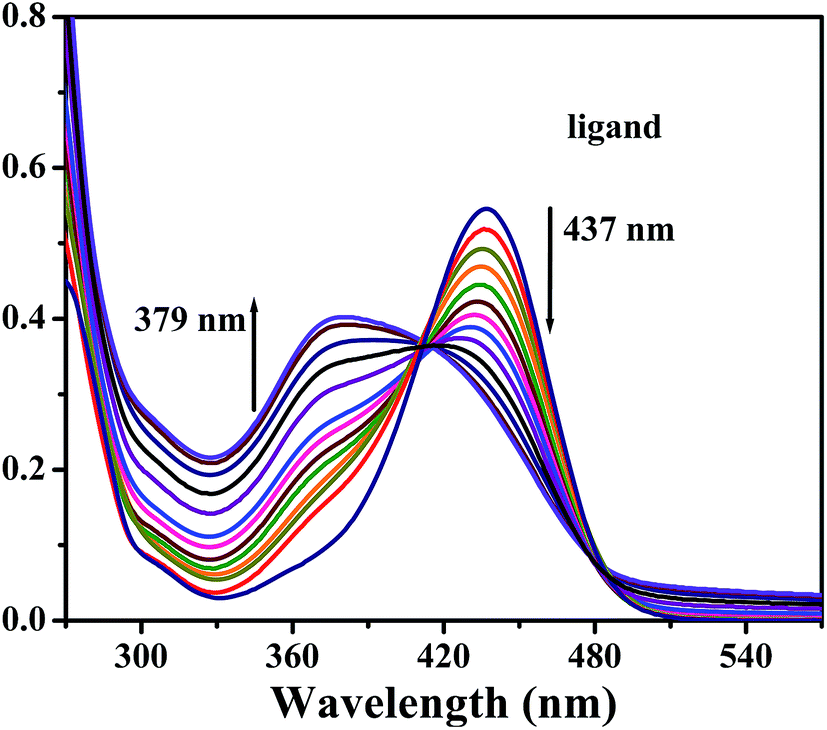
The electronic spectrum of the ligand (H2L) comprises of a pronounced band at 437 nm (ε = 10900 M−1 cm−1) in 1:1 water–acetonitrile solution (see Fig. 10) which undergoes a blue shift on addition of zinc nitrate to 379 nm (ε = 8000 M−1 cm−1). Few examples of ratiometric response of some ligands to metal ions with a distinct blue shifted emission were reported previously.34 In our case, may be due to macrocyclization of the ligand H2L, a stained H2L′ was formed that inhibits the internal charge transfer, causing blue shift. H2L hardly exhibits fluorescence (yellowish green) emission at 523 nm in 1:1 water–acetonitrile upon excitation at 437 nm (Fig. 11). Now zinc nitrate solution in 50:50 water–acetonitrile (0–65 μM) was added to the solution of H2L and fluorescence titration was performed. A new fluorescence emission peak at about 480 nm appeared and the intensity was slowly enhanced, which may consider as a signature of a Zn2+ selective ratiometric fluorescent signaling behavior. The rapid decrease in emission intensity at 523 nm is probably due to the coordination of phenolic oxygen to Zn2+ ion and the slow appearance of emission peak at 480 nm can be ascribed to zinc-ion binding to the partially hydrolyzed ligand (H2L′) formed in situ via hydrolysis of a CN bond. Fig. 12 shows fluorescence photographs of ligand (H2L) and ligand (H2L) + zinc nitrate under UV lamp excited at 365 nm. We have also verified using UV spectra that no hydrolysis of ligand has been taken place without zinc ion (Fig. S10, ESI†). The quantum yield is 0.231 for ligand only. However, it decreases when same amount of Zn2+ is added to the ligand H2L and the quantum yield value is 0.145. This may be due to the quenching effect upon addition of zinc ion to ligand.
 |
| | Fig. 10 Spectrophotometric titration of H2L (0.5 × 10−4 M) with zinc nitrate solution (0–1 × 10−4 M) in 50:50 water–acetonitrile. | |
 |
| | Fig. 11 Fluorescence titration of H2L (50 μM) in presence of increasing concentrations of zinc nitrate (0–65 μM) in 50:50 water–acetonitrile. | |
 |
| | Fig. 12 Fluorescence photographs of H2L and H2L + Zn(NO3)2 under UV lamp excited at 365 nm. | |
Selectivity of H2L towards ZnII
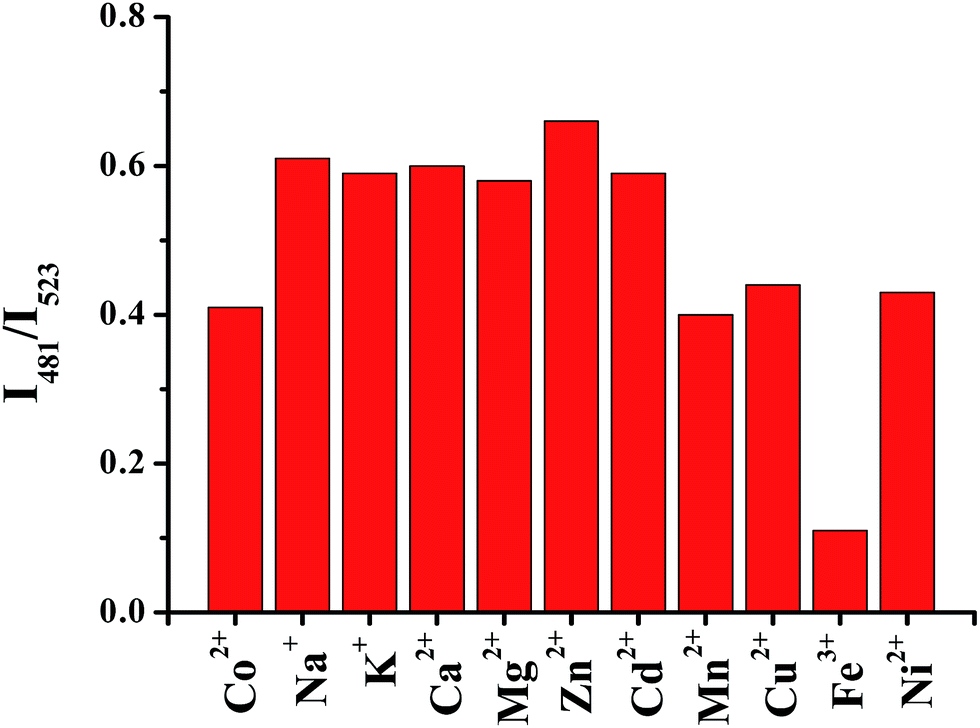
In coordination studies with other biologically relevant metal cations, ligand (H2L) shows a selective response towards ZnII. Since coordination of ZnII results in a blue shift of the emission maximum to 481 nm, the emission intensity at this wavelength is a good indicator for the selectivity of the probe response. With up to milli-molar concentrations of CaII, MgII, Na+ and K+ the emission spectra of the H2L slightly enhanced, whereas the paramagnetic metal cations CuII, FeII, NiII, CoII, and MnII, resulted in fluorescence quenching (Fig. 13a). We also investigated the emission response for CdII which is a d10 metal cation exhibiting similar coordination properties like ZnII. In the presence of CdII, H2L shows quenching effect in emission intensities at 523 nm. Fig. 13b shows the emission intensity ratios at 481 and 523 nm as a function of selected divalent metal cations (red columns). Since fluorescence emission band at 523 nm in the presence of cations other than ZnII is not shifted, it should be possible to distinguish the fluorescence ratio at 481 and 523 nm from the ZnII-selective emission change. To further justify the selectivity of the ligand H2L, the emission response of each cation was measured in the presence of an equimolar amount of ZnII (see Fig. 14) in 1:1 water–acetonitrile. Under this condition the emission ratio for FeII shows a lower value compared to that of the other cations. In present study, we are reporting the chemodosimetric property of H2L towards metal ions. Sensitivity of H2L towards anions is underway in our laboratory, will be published soon.
 |
| | Fig. 13 (a) Fluorescence responses of H2L (50 μM) in the presence of various analytes (50 μM); (b) fluorescence ratio (I481/I523) of ligand (50 μM) in the presence of various analytes (50 μM) in 50:50 water–acetonitrile. | |
 |
| | Fig. 14 Fluorescence ratio (I481/I523) of H2L (50 μM) in presence of Zn2+ and individual analyte in equal concentration (50 μM) in 50:50 water–acetonitrile. | |
Experimental section
Materials and physical methods
All reagents and chemicals were purchased from Sigma and used without further purification. Solvents used for spectroscopic studies were purified and dried by standard procedures before use. UV-Vis titration was recorded on SHIMADZU UV-2450 spectrophotometer. All the solutions were prepared in acetonitrile solvent of UV-Vis spectroscopy grade. The concentration of solutions used for UV-Vis studies was kept in the range of ∼10−4 to 10−5 M in order to avoid precipitation. Infrared (IR) samples were prepared as KBr pellets, and spectra were obtained in the 400–4000 cm−1 range using a Perkin-Elmer 1600 FTIR spectrometer. Elemental analyses were performed on a Perkin-Elmer Model 2400 analyzer. 1H NMR data were collected using an AM-300 spectrometer. Chemical shifts have been reported in δ relative to TMS. Emission spectra were recorded by Perkin Elmer LS55 fluorescence spectrophotometer equipped with a 10 mm quartz cell and a thermostat bath at 298 K temperature after subtraction of solvent background. The electrospray mass spectra were recorded on a MICROMASS Q-TOF mass spectrometer. All emission spectral measurements were done with ∼10−6 to 10−7 M concentration range of solute in order to avoid aggregation and self quenching.
Syntheses of ligand and complexes
Ligand (H2L). A solution containing 1 mmol of 1,3-propane diammine (0.078 g) in 10 mL ethanol was added dropwise to a solution of 1 mmol (0.189 g) of 2,6-diformyl-4-tert-butylphenol in 20 mL ethanol. An intense yellow solid precipitated. After filtration the solid was washed several times with ethanol. The solid compound was dried under CaCl2 desiccator and sample purity was monitored by TLC. Yield-80%; M.W.-450; 1H NMR (300 MHz, d6-DMSO, 25 °C, TMS): δ = 8.53 (2H, s); 7.67 (2H, s); 7.31 (2H, dd, J = 5.1 and J = 1.2 Hz); 6.89–6.94 (4H, m); 3.83 (4H, t, J = 6.6 Hz); 3.16 (4H, t, J = 6.6 Hz); FT-IR (KBr pellet, cm−1): 2926 (m), 2858 (s), 1682.5 (s), 1633.1 (s), 1599.7 (m), 1462 (s), 1301 (s), 1254 (s), 969 (s), 633 (s).
Dinuclear macrocyclic zinc(II) complex, [Zn2(L′)(NO3)2] (1). To a water–acetonitrile suspension of H2L (1 mmol, 0.450 g), a solution of zinc nitrate hexahydrate (1 mmol, 0.297 g) in 10 mL water–acetonitrile was added slowly. The resulting solution was stirred for 20 min and a yellow solution was obtained. After keeping the solution for two days needled shaped yellow crystals suitable for X-ray structural determination were separated out. Yield-84%; M.W.-741.40; elemental analysis (%) calculated for C30H38N6O8Zn2: C, 48.55; H, 5.12; N, 11.32; found: C, 48.52; H, 5.11; N, 11.31.
Dinuclear side-off copper(II) complex, [Cu2(L)(NO3)2] (2). Complex 2 was prepared by following similar procedure as used for complex 1. Here copper nitrate hexahydrate (1 mmol, 0.297 g) was used in place of zinc nitrate hexahydrate. The resulting solution was stirred for 45 min and a green solution was obtained. After keeping the solution for 2 days block shaped deep crystals suitable for X-ray structural determination were separated out. Yield-89%; M.W.-699.64; elemental analysis (%) calculated for C27H32Cu2N4O10: C, 46.30; H, 4.57; N, 8.01; found: C, 46.28; H, 4.53; N, 7.99.
Dinuclear side-off nickel(II) complex, [Ni2(L)(NO3)(H2O)3] (3). Complex 3 was synthesised by following the same procedure as reported earlier.35
X-Ray data collections and structure determinations
Diffraction data for compounds 1, 2 and 3 were collected on a Bruker Smart Apex diffractometer equipped with CCD. All the experiments were performed at room temperature with Mo-Kα radiation (λ = 0.71073 Å). Cell refinement, indexing and scaling of the data set were carried out using Bruker Smart Apex and Bruker Saint Packages.36 The structures were solved by direct methods and subsequent Fourier analyses and refined by the full-matrix least-squares method based on F2 with all observed reflections.37 In complex 2 atoms C3, C4, C6 and t-But group disordered over two positions with refined occupancies of 0.56(1)/0.44(1). The contribution of H atoms (at calculated position generated by program SHELXL)37 was introduced in the final cycles of refinement. Crystallographic data and details of refinements are reported in Table 3. Complex 3 was previously reported.35 All the calculations were performed using the WinGX System, Ver 2013.3.38
Table 3 Crystallographic data and details of refinement for complexes 1 and 2
| |
1 |
2 |
| R1 = ∑||Fo| − |Fc||/∑|Fo|, wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2. |
| Empirical formula |
C30H38N6O8Zn2 |
C27H32Cu2N4O10 |
| fw |
741.40 |
699.64 |
| System |
Monoclinic |
Monoclinic |
| Space group |
P21/n (no. 14) |
C2 (no. 5) |
| a (Å) |
7.1937(15) |
12.2643(7) |
| b (Å) |
19.343(4) |
10.4917(6) |
| c (Å) |
11.985(3) |
12.4618(7) |
| α (deg) |
90 |
90 |
| β (deg) |
103.105(9) |
113.610(2) |
| γ (deg) |
90 |
90 |
| V (Å3) |
1624.3(6) |
1469.28(15) |
| Z |
2 |
2 |
| Dcalcd (g cm−3) |
1.516 |
1.581 |
| μ (mm−1) |
1.535 |
1.510 |
| F(000) |
768 |
720 |
| θ range, deg |
2.04–22.26 |
1.8–27.1 |
| Total reflns |
14895 |
9150 |
| Unique reflns |
2055 |
2983 |
| Rint |
0.054 |
0.031 |
| Reflns (I > 2σ(I)) |
1753 |
2549 |
| Refined parameters |
208 |
200 |
| Goodness-of-fit (F2) |
1.093 |
1.069 |
| R1, wR2 (I > 2σ(I))a |
0.0309, 0.0816 |
0.0396, 0.1047 |
| Residuals (e Å−3) |
−0.22, 0.32 |
−0.30, 0.60 |
Conclusions
The treatment of zinc(II) nitrate with N,N′-propylenebis(3-formyl-5-tert-butylsalicylaldimine), an acyclic side-off compartmental ligand (H2L) produces a dinuclear macrocyclic zinc(II) complex [Zn2L′(NO3)2], where the macrocyclic ligand H2L′ is formed due to ZnII catalyzed hydrolysis of the starting ligand H2L. Interestingly, the H2L in presence of nickel(II) and copper(II) nitrate forms dinuclear metal complexes, [Ni2(L)(NO3)(H2O)3] and [Cu2(L)(NO3)2] where starting ligand remains unchanged. The formation of macrocyclic ligand H2L′ in solution has been authenticated by several spectroscopic means. The ligand H2L is highly fluorescent and, its fluorescence intensity is quenched in presence of other metal ions other than ZnII. In presence of ZnII a shift in emission band is observed and thus, the ligand (H2L) acts as ratiometric chemodosimeter for the selective detection of zinc(II) ion in solution. Furthermore, the macrocyclization process is very fast and exclusively Zn2+ sensitive. Hence, this work demonstrates an inexpensive way for the selective detection of Zn2+ ion at nearly 50 μM level concentration in a chemodosimetric way in presence and/or absence of other metal ions.
Acknowledgements
The authors wish to thank the Council of Scientific and Industrial Research (CSIR), New Delhi [project number 01(2464)/11/EMR-II dated 16-05-2011, to D. D. and project number 09/028(0766)/2010-EMR-I dated 22/02/2010, to S. D.] for financial support. The authors also thankful the Department of Science and Technology (DST), New Delhi for providing the single crystal diffractometer facility at the Department of Chemistry, University of Calcutta, through the DST-FIST program. We are grateful to Professor Ennio Zangrando, Department of Chemical and Pharmaceutical Sciences, University of Trieste, Via L. Giorgieri 1, 34127 Trieste, Italy for his kind help to solve the single crystal X-ray diffraction data.
References
- M. Y. Chae and A. W. Czarnik, J. Am. Chem. Soc., 1992, 114, 9704–9705 CrossRef CAS
 .
. - K. Kaura, R. Sainia, A. Kumara, V. Luxamib, N. Kaurc, P. Singha and S. Kumara, Coord. Chem. Rev., 2012, 256, 1992–2028 CrossRef .
- Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2013, 113, 192–270 CrossRef CAS PubMed .
- D. T. Quang and J. S. Kim, Chem. Rev., 2010, 110, 6280–6301 CrossRef CAS PubMed .
- J. Du, M. Hu, J. Fan and X. Peng, Chem. Soc. Rev., 2012, 41, 4511–4535 RSC .
-
(a) K. Mariappan, M. Alaparthi, G. Caple, V. Balasubramanian, M. M. Hoffman, M. Hudspeth and A. G. Sykes, Inorg. Chem., 2014, 53, 2953–2962 CrossRef CAS PubMed ;
(b) X. Lu, Z. Guo, M. Feng and W. Zhu, ACS Appl. Mater. Interfaces, 2012, 4, 3657–3662 CrossRef CAS PubMed ;
(c) M. H. Lee, S. W. Lee, S. H. Kim, C. Kang and J. S. Kim, Org. Lett., 2009, 11(10), 2101–2104 CrossRef CAS PubMed .
-
(a) B. N. Ahamed, M. Arunachalam and P. Ghosh, Inorg. Chem., 2010, 49, 4447–4457 CrossRef CAS PubMed ;
(b) N. C. Lim, S. V. Pavlova and C. Brückner, Inorg. Chem., 2009, 48, 1173–1182 CrossRef CAS PubMed ;
(c) Y. Liu, M. Li, Q. Zhao, H. Wu, K. Huang and F. Li, Inorg. Chem., 2011, 50, 5969–5977 CrossRef CAS PubMed .
-
(a) G. Ajayakumar, K. Sreenath and K. R. Gopidas, Dalton Trans., 2009, 1180–1186 RSC ;
(b) H. Zhang, L. Xie, W. Yan, C. He, X. Cao and C. Duan, New J. Chem., 2009, 33, 1478–1481 RSC ;
(c) A. Basu and G. Das, Dalton Trans., 2011, 40, 2837–2843 RSC .
-
(a) J. Liu, C. Li and F. Li, J. Mater. Chem., 2011, 21, 7175–7181 RSC ;
(b) Q. Zou, L. Zou and H. Tian, J. Mater. Chem., 2011, 21, 14441–14447 RSC ;
(c) M. J. Culzoni, A. M. de la Peña, A. Machuca, H. C. Goicoechea and R. Babianoc, Anal. Methods, 2013, 5, 30–49 RSC .
-
(a) J. Li, Y. Zeng, Q. Hu, X. Yu, J. Guo and Z. Pan, Dalton Trans., 2012, 41, 3623–3626 RSC ;
(b) X. Zhang, Y. Xu, P. Guo and X. Qian, New J. Chem., 2012, 36, 1621–1625 RSC ;
(c) W. T. Gong, B. Gao, J. Z. Zhao and G. L. Ning, J. Mater. Chem. A, 2013, 1, 5501–5504 RSC ;
(d) Q. Wang, C. Li, Y. Zou, H. Wang, T. Yi and C. Huang, Org. Biomol. Chem., 2012, 10, 6740–6746 RSC .
- L. Wang, M. Yu, Z. Liu, W. Zhao, Z. Li, Z. Ni, C. Li and L. Wei, New J. Chem., 2012, 36, 2176–2179 RSC .
- R. K. Jackson, Y. Shi, X. Yao and S. C. Burdette, Dalton Trans., 2010, 39, 4155–4161 RSC .
-
(a) M. P. Cuajungco and K. Y. Faget, Brain Res. Rev., 2003, 41, 44 CrossRef CAS PubMed ;
(b) M. Ebadi, F. Perini, K. Mountjoy and J. S. Garvey, J. Neurochem., 1996, 66, 2121 CrossRef CAS PubMed ;
(c) J. M. Berg and Y. Shi, Science, 1996, 271, 1081 CAS ;
(d) B. L. Vallee and K. H. Falchuk, Physiol. Rev., 1993, 73, 79–118 CAS ;
(e) C. J. Frederickson, Int. Rev. Neurobiol., 1989, 31, 145–238 CrossRef CAS PubMed ;
(f) D. W. Choi and J. Y. Koh, Annu. Rev. Neurosci., 1998, 21, 347–375 CrossRef CAS PubMed .
-
(a) G. R. Rout and P. Das, Agronomie, 2003, 23(1), 3–11 CrossRef ;
(b) S. e. Smith and E. J. Larson, J. Biol. Chem., 1946, 163, 29–38 CAS ;
(c) B. T. A. Muyssen, S. De, A. C. Karel and C. R. Janssen, Aquat. Toxicol., 2006, 77(4), 393–401 CrossRef CAS PubMed .
- Z. Li, M. Yu, L. Zhang, M. Yu, J. Liu, L. Wei and H. Zhang, Chem. Commun., 2010, 46, 7169–7171 RSC .
- C. E. Felton, L. P. Harding, J. E. Jones, B. M. Kariuki, S. J. A. Pope and C. R. Rice, Chem. Commun., 2008, 6185–6187 RSC .
-
(a) R. Y. Tsien and A. T. Harootunian, Cell Calcium, 1990, 11, 93 CrossRef CAS PubMed ;
(b) A. Coskun and E. U. Akkaya, J. Am. Chem. Soc., 2005, 127, 10464 CrossRef CAS PubMed ;
(c) A. P. Demchenko, J. Fluoresc., 2010, 20, 1099 CrossRef PubMed ;
(d) Y. Qu, J. Hua and H. Tian, Org. Lett., 2010, 12, 3320 CrossRef CAS PubMed .
- G. Grynkiewicz, M. Poenie and R. Y. Tsien, J. Biol. Chem., 1985, 260, 3440 CAS .
- M. M. Yu, Z. X. Li, L. H. Wei, D. H. Wei and M. S. Tang, Org. Lett., 2008, 10(22), 5115–5118 CrossRef CAS PubMed .
- P. A. Vigato and S. Tamburini, Coord. Chem. Rev., 2008, 252, 1871–1995 CrossRef CAS .
- P. A. Vigato and S. Tamburini, Coord. Chem. Rev., 2004, 248, 1717–2128 CrossRef CAS .
- P. A. Vigato, U. Casellato and D. E. Fenton, Coord. Chem. Rev., 1990, 106, 25–170 CrossRef CAS .
- A. J. Atkins, D. Blake, A. Marin-Bederra, S. Parsons, L. Ruiz-Ramirez and M. Schröder, Chem. Commun., 1996, 457–464 RSC .
- H. Okawa, H. Furutachi and D. E. Fenton, Coord. Chem. Rev., 1998, 174, 51–75 CrossRef CAS .
- M. Suzuki, H. Furutachi and H. Okawa, Coord. Chem. Rev., 2000, 200–202, 105–129 CrossRef CAS .
- B. Dutta, P. Bag, U. Florke and K. Nag, Inorg. Chem., 2005, 44, 147–157 CrossRef CAS PubMed .
- S. Hazra, S. Majumder, M. Fleck, R. Koner and S. Mohanta, Polyhedron, 2009, 28(14), 2871–2878 CrossRef CAS .
- S. Majumder, L. Mandal and S. Mohanta, Inorg. Chem., 2012, 51(16), 8739–8749 CrossRef CAS PubMed .
- B. J. Hathway and G. Wilkinson, Comprehensive Coordination Chemistry, John Wiley & Sons, New York, NY, USA, 1985 Search PubMed .
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier, Amsterdam, The Netherlands, 2nd edn, 1984 Search PubMed .
- J. Adhikary, A. Guha, T. Chattopadhyay and D. Das, Inorg. Chim. Acta, 2013, 406, 1–9 CrossRef CAS .
- R. R. Gagne and A. K. Shiemke, J. Am. Chem. Soc., 1981, 103, 4073–4081 CrossRef CAS .
-
(a) P. Job, Ann. Chim. Appl., 1928, 9, 113–203 CAS ;
(b) Z. D. Hill and P. MacCarthy, J. Chem. Educ., 1986, 63(2), 162 CrossRef CAS .
-
(a) H. J. Kim, S. J. Lee, S. Y. Park, J. H. Jung and J. S. Kim, Adv. Mater., 2008, 20, 3229–3234 CrossRef CAS ;
(b) L. Tang, M. Cai, P. Zhou, J. Zhao, K. Zhong, S. Hou and Y. Bian, RSC Adv., 2013, 3, 16802–16809 RSC ;
(c) X. Peng, J. Du, J. Fan, J. Wang, Y. Wu, J. Zhao, S. Sun and T. Xu, J. Am. Chem. Soc., 2007, 129, 1500–1501 CrossRef CAS PubMed ;
(d) Y. Zhao, J. Sun, Z. Shi, C. Pan and M. Xu, Luminescence, 2011, 26, 214–217 CrossRef CAS PubMed .
- S. Das, P. Maiti, T. Ghosh, E. Zangrando and D. Das, Inorg. Chem. Commun., 2012, 15, 266–268 CrossRef CAS .
- SMART, SAINT, Software Reference Manual, Bruker AXS Inc., Madison, WI, 2000 Search PubMed .
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed .
- L. Farrugia, WinGX System, Ver2013.3, University of Glasgow Search PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1410679 and 1410680 contain the supplementary crystallographic data for complexes 1. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra05478a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.