
Self-expansion, self-exfoliation and self-dispersion: insights into colloidal formation of atomically thin two-dimensional MoO2.5(OH)0.5†
Hai Wang*ab and
Yan Sub
aKey Laboratory of New Processing Technology for Nonferrous Metals and Materials, Ministry of Education, Guilin University of Technology, Guilin 541004, China. E-mail: hbwanghai@gmail.com; Fax: +86-773-5896671; Tel: +86-773-5896672
bDepartment of Electromechanical Engineering, FST, University of Macau, Taipa, Macau
First published on 7th October 2016
Abstract
Large-scale synthesis of colloidal two-dimensional (2D) monolayered nanosheets has been a great challenge for material science. The difficulties are caused by the low exfoliation ability of 2D bulk materials, the poor dispersion and high instability of colloidal solutions. Conventional exfoliation and dispersion of these bulk 2D materials usually involve chemical modification via functionalization, intercalation or oxidation, which was detrimental to their intrinsic physical and chemical properties. In the present study, we develope a novel “green” liquid exfoliation method to prepare highly dispersible colloidal MoO2.5(OH)0.5 nanosheets in water. The as-obtained water-dispersible colloidal solutions (average particle size 20 nm and thickness 0.6–0.8 nm) kept in stable state longer than 10 months and show time-dependent photoluminescence (PL). Based on the results of combined time-dependent ex situ TEM observation experiments and ex situ XRD simulations, a novel “self-expansion and self-exfoliation” model was proposed to elucidate the formation of colloidal formation of atomically thin 2D MoO2.5(OH)0.5. It is expected that the present method has potential to be extended to colloidal formation of other layered compounds.
Introduction
Encouraged by the discovery of graphene, two-dimensional (2D) monolayered nanostructures, including h-BN and transition-metal dichalcogenides (TMD) nanosheets attracted considerable attention, because of their unique structure and fascinating physicochemical properties comparing to the bulk counterpart.1–4 These monolayered nanosheets can be utilized as the fundamental building blocks for constructing multifunctional nanostructures.5–7 Recently, a lot of efforts have been made to prepare 2D colloidal nanocrystals for applications in optical, electrical, mechanical fields (partially summarized in Table S1†).1–4,8–20 However, how to synthesise colloidal nanocrystals with atomic-level thickness is still a great challenge, due to the low exfoliation ability of 2D bulk materials, poor dispersion and high instability of their colloidal solutions. To the best of our knowledge, there are limited numbers of reports on the preparation of colloidal nanocrystals with atomic-level thickness.21–24 This may be due to the fact that TMD (MoS2, ReS2, TiS2, TaS2, and GaSe) are easily exfoliated by organic solvents.21–24 The prior referred synthetic methods did not focused on layered oxides, although they indicated some possibilities in preparing the monolayered 2D colloidal solutions, such as graphene, g-C3N4, h-BN and TMD.1–4,8–20The early research on the molybdenum oxide hydroxide MoO2.5(OH)0.5 can be traced back to O. Glemser's work in 1951.25 The synthesis routes of MoO2.5(OH)0.5 had been extensive studied in the early stage.26,27 However, prior researchers did not use it for 2D nanosheets colloidal solutions, in spite of its similar structure as its parent oxide MoO3. The possibility of its exfoliation hasn't been explored due to the lack of effective methods, and even fewer study on its stability of its colloidal solutions.
Due to the unique stacked structure, weakening the van der Waals interactions between adjacent layers is essential for exfoliation of MoO2.5(OH)0.5. Currently, the effective exfoliation process from bulk layered materials was restricted to chemical modification via functionalization, intercalation or oxidation.1–4,8–20 These chemical reactions will introduce numerous impurities or defects to the parent materials, and will severely change or degrade the intrinsic properties of the resulting materials.3,9,14,17,19,28–30 On the other hand, to enhance the exfoliation of 2D colloidal solutions, ultrasonic processes with scalable features are implemented. However, ultrasonic treatment tend to cause aggregation, which need proper surfactants.31–33 Hence, it is highly desirable to develop efficient alternative methods of 2D colloidal solutions based on MoO2.5(OH)0.5.
Recent success in direct exfoliation and dispersion of 2D materials (graphene, h-BN, MoS2, WS2 and MoSe2) in pure water via temperature control makes it feasible to simultaneously exfoliate and disperse 2D MoO2.5(OH)0.5 colloidal nanocrystals in water.1 Bearing this in mind, we paid our attention to the comparative results of the lattice parameters and unit cell volume between orthorhombic MoO2.5(OH)0.5 and orthorhombic MoO3 (Scheme S1, Table S2 and ESI Note 1†). Compared to MoO3 crystals structure, two typical structural features of MoO2.5(OH)0.5 are favourable to the formation of colloidal solutions. One is the larger inter-layer distance of MoO2.5(OH)0.5, which makes the further exfoliation easier. The other is the OH group in MoO2.5(OH)0.5, which enhances the dispersion and stability of colloidal solutions.34,35
Previously, monolayered oxides has not been prepared successfully through direct exfoliation and dispersion of the bulk counterpart in water. In the present study, a novel “sauna reaction” method to prepare MoO2.5(OH)0.5 nanobelts directly from MoO3 nanobelts is developed based on the similarity of their crystal structure. The basic idea is to transform MoO3 into MoO2.5(OH)0.5 while maintaining their original MoO6 network structures. Experimental details are illustrated in Scheme 1. Compared with prior synthesis methods,26,27 the present method does not include other chemical reagents than H2O and fibrous cloth, so as to exclude the effects of organics on the formation of colloidal solutions. It is found that the fibrous cloth and high pressure are the two major factors for the synthesis of MoO2.5(OH)0.5 (Fig. S1 and ESI Note 2†).
 | ||
| Scheme 1 The schematic of synthesis process for MoO2.5(OH)0.5 nanobelts. (a) Schematic of “sauna reaction” within a confined space between two layers fibrous cloth. (b) Photos and schematic images of gray white raw MoO3 nanobelt powders and dark blue MoO2.5(OH)0.5 nanobelt powders. Carbon acted as a catalytic trigger for H to inset into the layer spacing of MoO3 nanobelts. | ||
Subsequently, we present a simple method for the direct exfoliation and dispersion of bulk MoO2.5(OH)0.5 nanobelts in pure water. Systematic ex situ experiments relying on XRD simulations and TEM observation were implemented to investigate the corresponding mechanism of the exfoliation. It is found that high stability of 2D MoO2.5(OH)0.5 nanosheets in water is maintained because of the presence of surface charges of nanosheets and OH group contained in the MoO2.5(OH)0.5.
Results and discussion
As illustrated in Scheme 1, the synthesis procedure consists of two main steps. First, the as-prepared MoO3 nanobelt powders are wrapped between two layers of fibrous cloth. Then, the whole parts are transferred to a Teflon-lined autoclave filled with a small amount of deionized water. More detailed descriptions are shown in Experimental section of ESI.†In order to validate the effectiveness of the proposed synthesis method of MoO2.5(OH)0.5 nanobelt powders, the crystal structure and microstructure of as-prepared dark blue powders are further characterized with XRD and FESEM, respectively. As shown in Fig. 1, all the diffraction peaks were ascribed to the MoO2.5(OH)0.5 phase with an orthorhombic structure (PDF#14-0041, a = 3.888 Å, b = 14.082 Å, and c = 3.734 Å). The results show that MoO3 powders had been completely transformed into MoO2.5(OH)0.5 powders by “sauna reaction” between two layers of fibrous cloth. Also shown in Fig. 1, the diffraction peaks of MoO2.5(OH)0.5 were similar to that of MoO3 besides the low-angle shift of typical peaks of (0l0). The expanded lattice distance of (0l0) created a favourable structural condition for further exfoliation of MoO2.5(OH)0.5 powders. The maximum diffraction intensity of as-transformed powders happened at (040). This obviously shows that (0l0) planes had the highly preferred orientation. Meanwhile, the other diffraction peaks of MoO2.5(OH)0.5 shifted to a lower diffraction angle compared to those of MoO3. Hence, the distorted octahedral layers of MoO6 were slightly changed from the pristine MoO3 during the conversion process. This phenomenon was consistent with previous studies.36,37
 | ||
| Fig. 1 The XRD patterns of MoO3 nanobelts and MoO2.5(OH)0.5 nanobelts. The corresponding powders are shown in the inset (a), schematic illustration showing the structural relationships between layered orthorhombic MoO3 and layered orthorhombic MoO2.5(OH)0.5 (b). | ||
Furthermore, the OH surface group of dark blue MoO2.5(OH)0.5 powders (inset of Fig. 1) was evidenced by XPS measurements (Fig. S2†).38 It proves that the reaction was similar to hydrogen injection into the bulk MoO3. Similarly, the process of H insertion into transition metal oxides has been investigated by some research groups.39–43 It is also observed that the morphologies of pristine MoO3 powders were unchanged while keeping the pristine nanobelts stable, as shown in Fig. S3a and b.† This indicates a typical topotactic transformation of MoO3 to MoO2.5(OH)0.5.
Based on the above analysis, it is evidently shown that MoO2.5(OH)0.5 could be successfully obtained by the unique “sauna reaction”. The whole reaction does not involve with organic compounds or other surfactants. The proposed method provided more favourable conditions for investigating the formation and mechanism of stable colloidal solutions comparing to prior methods.26,27
A surprising phenomenon was observed, when the as-prepared MoO2.5(OH)0.5 powders were directly poured into water. It is amazing to observe that the powders dispersed instantly in water without any other treatment. The obtained colloid solutions looked like ink. On the contrary, if pristine MoO3 powders were added into water, the resulting solutions were in a suspension state. For MoO2.5(OH)0.5 colloidal solutions, an obvious Tyndall effect was observed when irradiated with a laser beam. This indicates the presence of colloidal nanoparticles (Fig. S4†). This phenomenon is unusual, owing to a well-known fact that pristine MoO3 nanobelt powders were insoluble in water.26 We also verified that MoO3 powders are not soluble in water, regardless of the water temperature. If MoO3 is dissolved in water, it is impossible to obtain MoO3 powders under hydrothermal reaction conditions. Generally speaking, the size of colloidal nanoparticles is below 100 nm. Because the dimension of single MoO2.5(OH)0.5 nanobelt is similar to that of single MoO3 nanobelt (Fig. S3a and b†), the Tyndall effect motives us to find out how colloidal nanoparticles are formed from MoO2.5(OH)0.5 nanobelts. Through the ex situ TEM observation (Fig. 2a and b), nanosheets traces together with some fragments were found in the colloidal solutions at the initial stage. Obviously, MoO2.5(OH)0.5 nanobelts were partially destroyed at the early stage. It is also found that the fingers were parallel to each other, which are similar to crystalline lattice fringes (Fig. 2b and c). It can be inferred that this result was attributed to the crystal lattice expansion of MoO2.5(OH)0.5.
 | ||
| Fig. 2 The evolution process of aqueous dispersions of monolayered MoO3 nanosheets. (a) and (b) TEM images of MoO2.5(OH)0.5 nanosheets instantly dispersed in water at initial stage. Some ultrathin sheets were packed together. (c) The schematic of the red rectangle in (b). (d)–(j) TEM images of MoO2.5(OH)0.5 nanosheets dispersed in water for different stages. (d)–(f) After ca. 1 month, (g)–(i) after ca. 4 months, (j) after ca. 8 months. (k) Tapping mode AFM images of the colloidal solutions deposited on a mica substrate. The corresponding height profiles indicate that the thickness of nanosheets was 0.6–0.8 nm (l), indicating the presence of monolayered MoO2.5(OH)0.5 nanosheets. | ||
Another unusual phenomenon is observed after the solutions were stood for ca. 8 months. The initial dark blue colloidal solutions were completely evolved into stable yellow ones. This evolution was a slow and gradual process (Movie S1†). The transformed supernatants are colloidal solutions, as proven by observation of the Tyndall effect with a handheld laser pointer. Then, we carried out a preliminary TEM observation for the yellow colloid solutions. As shown in Fig. S5,† the colloidal solutions comprised a large amount of colloidal particles with the uniform size of ca. 20 nm. Undoubtedly, the bulk single crystal MoO2.5(OH)0.5 nanobelts are completely exfoliated into highly water-dispersible colloidal particles. Moreover, the as-obtained colloidal aqueous solutions are highly stable up to over 10 months. To explain this unique phenomenon, it is necessary to reveal formation mechanism of colloidal nanoparticles.
As referred above, the final yellow colloidal solutions were formed over 8 months. Hence, we also designed a time-dependent experiment to track their clues using ex situ TEM measurements. We selected other three typical samples as comparisons, the blue colloidal solutions (1 month), the green colloidal solutions (after ca. 4 months) and the yellow colloidal solutions (after ca. 8 months).
As shown in Fig. 2d–f, the colloidal solutions (1 month) still contain some fragments. Different from its initial stage, none complete nanobelts are observed at this stage. Moreover, we found that some ultrathin nanosheets began to appear in some areas (Fig. S6†). Moreover, most of them were transformed into 3-dimensional (3D) continuous networks (Fig. 2d). This may due to the action of van der Waals interparticle forces based on the Doi and Edwards theory.44 Some broken and bending traces were clearly observed in Fig. 2f. By observation of both the fragments and the tearing nanosheets in Fig. S7,† we reasonably inferred that all nanosheets may be transformed into thinner fragments with time increasing.
When the evolution process lasted for ca. 4 months, we observed that 3D interconnected nanosheet networks still existed (Fig. 2g). Surprisingly, we found that some quantum dots-like were attached on the surface of those ultrathin sheets (Fig. 2g–i). A further question is that why quantum dots-like appeared on the surface of exfoliated ultrathin nanosheets. Actually, it was also found that the nanobelts began to become thinner and were evolved into some dots-like matters (Fig. 2d–f). It is obvious that these “dots” were from the evolution of the nanosheets, which maybe be attributed to the broken lattice. This will be further discussed in later section. Thus, we hypothesize that the remaining nanosheets would eventually be converted to quantum dot-like 2D materials.
With the continuous evolution process of colloidal solutions, after ca. 8 months, it is surprising to observe that pristine 3D networks (Fig. 2g–i) were completely evolved into uniform ultrathin nanosheets (Fig. 2j). The complete evolution confirmed our preliminary hypothesis. Moreover, the whole evolution process occurs only at room temperature without any treatment, such as sonication. To further investigate these detailed structural features of nanosheets-like particles, we enlarged the observation magnification. Unfortunately, the crystalline lattice patter from TEM is unstable at high-energy electron beams at 200 keV (Fig. S8†). A corresponding explanation is shown Fig. S8† below. To obtain the thickness and dimensions of as-evolved nanosheets, we performed atomic force microscope (AFM) measurements Fig. 2k and l represents tapping mode AFM scans of the nanosheets. The corresponding height profiles are shown in (Fig. 2l). The thickness of as-obtained nanosheets is ca. 0.6–0.8 nm with dimensions of ca. 20 nm. The thickness values suggest that these nanosheets are monolayer according to the crystal structure of MoO2.5(OH)0.5 (Fig. 3a). This is an encouraging result since there are no available literature record about the monolayered MoO2.5(OH)0.5 colloidal solutions. Noted that the resultant colloidal solutions are obtained without conventional sonication and centrifugation processes.
 | ||
| Fig. 3 The schematic illustration of the evolution process for preparation of the aqueous solutions (a and b), (c) the crystal structure of MoO2.5(OH)0.5. | ||
Based on time-dependent ex situ TEM experiments of colloidal solutions, we reasonably concluded that we have found a novel and effective strategy for preparation of monolayered MoO2.5(OH)0.5 nanosheets. As displayed in Fig. 3, the strategy for preparation of monolayered MoO2.5(OH)0.5 nanosheets consists of two steps: step (i) the exfoliation of as-obtained bulk MoO2.5(OH)0.5 nanobelts, and step (ii) the formation of 2D MoO2.5(OH)0.5 colloidal solutions. The as-obtained ultrathin nanosheets were eventually broken into monolayered MoO3 nanosheets with smaller thickness (Fig. 3b).
Two fundamental questions arise: (a) why could bulk single crystal nanobelts be eventually transformed into nanosheets with atomic-level thickness? (b) What's the corresponding formation mechanism of monolayered MoO2.5(OH)0.5 nanosheets? Maybe the bottom powders at the sample pools would provide us some valuable clues to answer these questions. All un-exfoliated bottom powders were poured off in previous large amounts of literature.45–47 Previous scientists did not paid enough attention to un-exfoliated powders in the colloidal solutions.
We carefully collected the un-exfoliated powders from the bottom of the colloidal solutions. Subsequently, we performed time-dependent ex situ XRD tracing experiments simulation. Interestingly, we found that the crystal phase of un-exfoliated powders in the initial stage was similar to as-prepared pristine MoO2.5(OH)0.5 without obvious changes (Fig. 4a). When the powders were soaked for ca. 20 days, we found that most peak positions were nearly the same as the initial ones. The relative intensity of some diffraction peaks, such as, (110), (0l0), (021), (130), (111) became stronger, while the relative intensities of (020), (040) and (060) was nearly unchanged when compared to that of pristine MoO2.5(OH)0.5. With the time prolonged after 30 days, the positions of the (0l0) reflections in the XRD patterns were shifted to a higher d value. This indicates a lattice expansion in a direction normal to the (0l0) planes. At this stage, nearly all peaks shifted, especially (020) and (040) peaks. Moreover, some MoO3 peaks began to reappear (after 30 days). The interlayer spacing of MoO2.5(OH)0.5 crystal structure for different periods is schematically shown in Fig. 4b. All these results provided us two important evidences. First, the layer distance was expanded without any treatment, which may be caused by H2O molecules into (0l0) layers of MoO2.5(OH)0.5.48,49 Second, the structural reconstruction for the distorted MoO6 octahedral layer occurred, because some MoO3 diffraction peaks appeared, and some peak intensities changed. Similar phenomenon including the lower angle shift was observed at the 40th days and the 30th days after the bottom powders continued to immerse.
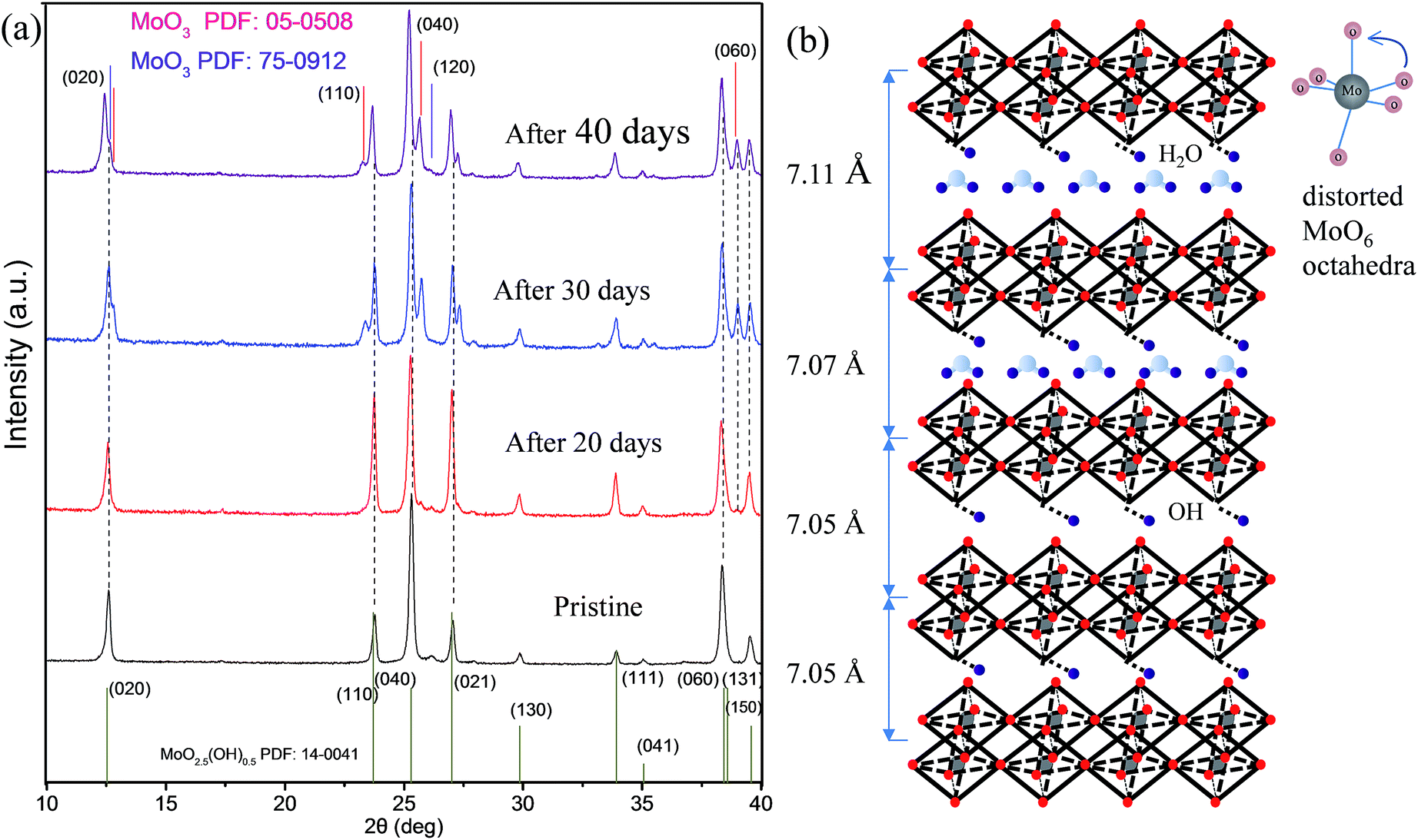 | ||
| Fig. 4 (a) The comparison of XRD patterns of MoO2.5(OH)0.5 nanobelts for different periods, pristine, after 20 days, 30 days and 40 days, (b) the corresponding schematic illustration of changes of the layer spacing of MoO2.5(OH)0.5 crystal structure for different periods in (a). The MoO6 octahedra reconstruction (blues) to accommodate the structural deformation thus resulted from the incorporation of OH group and the insertion of H2O molecular. | ||
As time increases, the value of d(020) increased from 7.05 to 7.11 Å. The spacing of layers d(020) was calculated by use of Scherrer's equation. The result shows that it was a continuous self-expansion process and H2O molecular gradually inserted into (0l0) layers of MoO2.5(OH)0.5. This is identical to the insertion of H2O molecular in LDH.50,51 A corresponding discussion is shown in Scheme S2 and ESI Note 3.† Based on ex situ time-dependent TEM observations and XRD tracing experiments, we reasonably draw a conclusion that the whole process was also a self-exfoliation process. All these evidences indicate that the self-exfoliation process was accompanied by a self-expansion process. This process is somewhat similar to natural exfoliation of deadwood immersed in a still water environment.
To answer the second question of whether MoO2.5(OH)0.5 powders could be directly transformed into 2D nanosheets in water, we must understand the formation of the fragments. Based on the XRD analysis at the 30th or 40th days after the preparation of the solvents (Fig. 4a), it is shown that the crystalline strain is a key factor of formation of fragments. As shown in Fig. 4a, the typical feature of the presence of lattice strain is obvious, because the XRD results of 30 days have the same peak shape and lower shift angles compared to those of 40 days.52 Fig. 4a also show that inside the double-layered distorted MoO6 octahedral, lower bonding strength may lead to destruction of monolayered MoO6 sheet layer. The lower bounding strength may be caused by changes of ionic and covalent bonding. Both the interweaving of the in-plane distortion and out-of-plane expansion would generate lattice strains. The interweaving lattice strains eventually led to the formation of fragments with atomic-scale thickness. Hence, the lattice strain generated during topochemical conversion is one of the key factors for the formation of the fragments. While weak van der Waals forces between double layers must be another key factor for formation of the fragment.
Then, why were the colloidal solutions highly stable during a long time? And why were they self-dispersible? As shown in Fig. S2,† OH group was contained in the MoO2.5(OH)0.5. This is different from conventional 2D colloidal solutions, such as, stable oxide graphene aqueous solutions and functionalized h-BN.35,53 In our case, the OH group was only from MoO2.5(OH)0.5 itself instead of from surfactants. Furthermore, we found that the as-formed yellow colloidal solutions were negatively charged. This was evidenced by the positively charged CTAB (Fig. S9a and b†) and zeta-potential measurements (Fig. S9c†). The negative charge may arise from substitution of Mo5+ for Mo6+ in MoO2.5(OH)0.5. Hence, the electrostatic stabilization maintains the highly stability of the colloidal solutions. The space resistance effects of organic surfactants can be excluded, because none any extra organic compound was introduced the synthesis process.
Furthermore, we explored the dispersal of MoO2.5(OH)0.5 nanobelt powders in other organic solvents. The used solvents included isopropanol (IPA), N-methyl-pyrrolidone (NMP), ethanol, and formamide. As shown in Fig. S10,† the formation of MoO2.5(OH)0.5 colloidal were much slower in the other solvents than in water. The results of our dispersion experiments demonstrated that water was the most effective for exfoliating bulk MoO2.5(OH)0.5 nanosheets into monolayered nanosheets. This may because the formation speed of monolayered nanosheets was dominated by the matching ability of the solvent and bulk materials in the surface energy levels. Previous studies also showed the matching of surface energy levels for the solvent and the bulk material was crucial for the formation of monolayered nanosheets.54 Based on this energy level matching requirement, fulfillment of potential energy levels without using any organic agents is extremely important for applications of aqueous colloidal solutions. It enables not only cost-effective future commercial applications but also facilitates investigation that needs aqueous colloidal solutions.
The above two-step experimental results as a “self-expansion and self-exfoliation” model is summarized in Scheme 2. This model clearly indicates the two-step formation mechanism of highly self-dispersible colloidal solutions of monolayered MoO2.5(OH)0.5 nanosheets. In the first “self-expansion” step, the MoO2.5(OH)0.5 nanobelts were first transformed from MoO3 nanobelts. This transformation was realized by a topochemical phase transformation reaction. During the transformation reaction, the H was inserted into MoO3 lattice, and the interlayer gap of MoO6 layers was increased. The as-prepared MoO2.5(OH)0.5 nanobelts exhibited a self-expansion phenomenon in water, because H2O molecular are inserted into between MoO6 layers. In the second “self-exfoliation” step, the as-prepared MoO2.5(OH)0.5 nanobelts were then transformed into monolayered nanosheets. The crystal strains caused by both the interweaving of the in-plane distortion, and out-of-plane expansion eventually led to the formation of nanosheets with thickness in atomic-level.
 | ||
| Scheme 2 Schematic of the formation mechanism of highly self-dispersible colloidal solutions of monolayered MoO2.5(OH)0.5 nanosheets by self-expansion and self-exfoliation process. The orthorhombic phase MoO3 nanobelts were transformed to the orthorhombic phase MoO2.5(OH)0.5 nanosheets by the “sauna reaction”. MoO2.5(OH)0.5 exhibited expansion of the interlayer distance by the topotactic phase strategies and H2O molecular intercalation (a). The combination mechanical behaviors of in-plane crystal strain and out-of-plane expansion led to the formation of fragments (b). | ||
To further explore other potential applications of MoO2.5(OH)0.5 colloidal solutions, it may be an opportunity for us to know their optical properties. As shown in Fig. 5a, the optical absorption spectra of three colloidal solutions exhibited a strong absorption band between 200–300 nm, which attributed to the charge transfer of the Mo–O band in the MoO6 octahedron.56 Based on original absorbance data of colloidal solutions, the corresponding optical band-gap energy (Eg) was obtained using a Tauc plot method. The band-gap values were found to be 3.42, 3.29 and 3.24 eV, respectively. Obviously, the Eg (3.42–3.24 eV) shows a red shift for the colloidal solutions, which may result from continuous exfoliation of nanoparticles in colloidal solutions. Generally speaking, the increase of band gap was attributed to the increase of crystalline degree and reduction of oxygen deficiency. In our case, the changes of colloidal solutions were a continuous self-exfoliation process. Thus, the crystalline degree of colloidal nanoparticles gradually became worse. It is expected that the tunable optical Eg of 2D colloidal solutions have potential applications as quantum dots colloidal solutions.
 | ||
| Fig. 5 (a) UV-vis absorption spectrum of the selected colloidal solutions, S1 (1 month), S2 (4 months) and S3 (8 months). The variation of (αhγ)1/2 with hγ is shown in the inset of (a), (b) photoluminescence emission spectra of the S1, S2 and S3 at 330 nm emission wavelengths. | ||
Photoluminescence (PL) can be used for investigating energy levels of 2D colloidal nanoparticles and can provide additional information of different stages of colloidal solutions.
The PL emission of three selected colloidal solutions (S1: 1 month, S2, 4 months, and S3, 8 months) was tested using 330 nm excitation wavelength at room temperature. As shown in Fig. 5b, the PL spectra presented two peaks position: a weak satellite peak at 373 nm and a moderate intensity peak at 446 nm. The PL peak position is consistent with MoO3 powders.55,56 The appearance of these peak positions were related to the imperfection of lattice and distort of MoO6 octahedra. The emissions at short wavelength excitation were believed to be the near band-edge transitions.55,56 The peak at 446 nm was obviously correlative with continuous exfoliation process of nanoparticles in colloidal solutions. This may because of the poorer crystalline degree and increased surface defects. The intensity of the peak at 446 nm decreased with reaction time. A large number of surface defects may exist on the nanosheets, owing to their high exposed surface area. Anyway, only PL data is insufficient to understand these new phenomena. Further research was necessary to obtain deeper insight into this interesting issue.
Conclusions
In conclusion, it is demonstrated that highly dispersible and stable colloidal MoO2.5(OH)0.5 nanosheets with monolayer-level thickness can form through direct self-exfoliation and self-dispersion of MoO2.5(OH)0.5 nanobelts in pure water. The larger interlayer distance of MoO2.5(OH)0.5 and its OH group determine whether the exfoliation and dispersion of MoO2.5(OH)0.5 can occur.A novel “self-expansion and self-exfoliation” model was built to reveal the mechanism of the transformation process from bulk single crystal MoO3 nanobelt powders to monolayered MoO2.5(OH)0.5 colloidal solutions based on the results of time-dependent ex situ TEM experiments and ex situ XRD simulations. It is found that the resulting colloidal MoO2.5(OH)0.5 nanosheets with monolayer-level thickness exhibited time-dependent photoluminescence.
This synthesis method of the demonstration model was unique because it avoided the introduction of exotic or organic compounds via a green “sauna reaction”. More importantly, this mechanism can shed some insights into future nanomaterial designs, especially for preparation of colloidal solutions of two-dimensional materials. We anticipate our present method could be extended to other layered compounds containing the OH functional group, such as Ni(OH)2, layered double hydroxides (LDH), and so on.
Acknowledgements
The authors thank Solar Energy Lab of University of Macau with projects FDCT/060/2014/A2 and the National Natural Science Foundation of China (No. 51462007) for financial support.Notes and references
- J. Kim, S. Kwon, D.-H. Cho, B. Kang, H. Kwon, Y. Kim, S. O. Park, G. Y. Jung, E. Shin and W.-G. Kim, Nat. Commun., 2015, 6, 8294 CrossRef CAS PubMed.
- W. Lei, V. N. Mochalin, D. Liu, S. Qin, Y. Gogotsi and Y. Chen, Nat. Commun., 2015, 6, 8849 CrossRef CAS PubMed.
- C.-J. Shih, A. Vijayaraghavan, R. Krishnan, R. Sharma, J.-H. Han, M.-H. Ham, Z. Jin, S. Lin, G. L. Paulus and N. F. Reuel, Nat. Nanotechnol., 2011, 6, 439–445 CrossRef CAS PubMed.
- N. D. Mansukhani, L. M. Guiney, P. J. Kim, Y. Zhao, D. Alducin, A. Ponce, E. Larios, M. J. Yacaman and M. C. Hersam, Small, 2016, 12, 294–300 CrossRef CAS PubMed.
- C. Altavilla, M. Sarno and P. Ciambelli, Chem. Mater., 2011, 23, 3879–3885 CrossRef CAS.
- H. Tang, K. P. Dou, C. C. Kaun, Q. Kuang and S. H. Yang, J. Mater. Chem. A, 2014, 2, 360–364 CAS.
- P. Sun, Q. Chen, X. Li, H. Liu, K. Wang, M. Zhong, J. Wei, D. Wu, R. Ma, T. Sasaki and H. Zhu, NPG Asia Mater., 2015, 7, e162 CrossRef CAS.
- S. K. Das, A. Bedar, A. Kannan and K. Jasuja, Sci. Rep., 2015, 5, 10522 CrossRef CAS PubMed.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS PubMed.
- X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2012, 135, 18–21 CrossRef PubMed.
- X. She, H. Xu, Y. Xu, J. Yan, J. Xia, L. Xu, Y. Song, Y. Jiang, Q. Zhang and H. Li, J. Mater. Chem. A, 2014, 2, 2563–2570 CAS.
- J. T. Han, J. I. Jang, H. Kim, J. Y. Hwang, H. K. Yoo, J. S. Woo, S. Choi, H. Y. Kim, H. J. Jeong and S. Y. Jeong, Sci. Rep., 2014, 4, 5133 Search PubMed.
- Y. Yao, L. Tolentino, Z. Yang, X. Song, W. Zhang, Y. Chen and C. P. Wong, Adv. Funct. Mater., 2013, 23, 3577–3583 CrossRef CAS.
- Z. Jin, J. R. Lomeda, B. K. Price, W. Lu, Y. Zhu and J. M. Tour, Chem. Mater., 2009, 21, 3045–3047 CrossRef CAS.
- A. Jawaid, D. Nepal, K. Park, M. Jespersen, A. Qualley, P. Mirau, L. F. Drummy and R. A. Vaia, Chem. Mater., 2015, 28, 337–348 CrossRef.
- A. O'Neill, U. Khan and J. N. Coleman, Chem. Mater., 2012, 24, 2414–2421 CrossRef.
- D. Li, M. B. Mueller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- U. Halim, C. R. Zheng, Y. Chen, Z. Lin, S. Jiang, R. Cheng, Y. Huang and X. Duan, Nat. Commun., 2013, 4, 2213 Search PubMed.
- Z. Zeng, Z. Yin, X. Huang, H. Li, Q. He, G. Lu, F. Boey and H. Zhang, Angew. Chem., Int. Ed., 2011, 50, 11093–11097 CrossRef CAS PubMed.
- X. Zhang, H. Wang, H. Wang, Q. Zhang, J. Xie, Y. Tian, J. Wang and Y. Xie, Adv. Mater., 2014, 26, 4438–4443 CrossRef CAS PubMed.
- H. Li, G. Lu, Z. Y. Yin, Q. Y. He, H. Li, Q. Zhang and H. Zhang, Small, 2012, 8, 682–686 CrossRef CAS PubMed.
- Q. N. Cui, J. Q. He, M. Z. Bellus, M. Mirzokarimov, T. Hofmann, H. Y. Chiu, M. Antonik, D. W. He, Y. S. Wang and H. Zhao, Small, 2015, 11, 5565–5571 CrossRef CAS PubMed.
- S. D. Lei, L. H. Ge, Z. Liu, S. Najmaei, G. Shi, G. You, J. Lou, R. Vajtai and P. M. Ajayan, Nano Lett., 2013, 13, 2777–2781 CrossRef CAS PubMed.
- Z. Y. Zeng, C. L. Tan, X. Huang, S. Y. Bao and H. Zhang, Energy Environ. Sci., 2014, 7, 797–803 CAS.
- O. Glemser and C. Naumann, Z. Anorg. Allg. Chem., 1951, 265, 288–302 CrossRef CAS.
- K. Eda, A. Sukejima, N. Matsuura and N. Sotani, Chem. Lett., 1998, 819–820 CrossRef CAS.
- R. L. Smith and G. S. Rohrer, J. Catal., 1998, 173, 219–228 CrossRef CAS.
- S. Stankovich, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Carbon, 2006, 44, 3342–3347 CrossRef CAS.
- C. Gomez-Navarro, J. C. Meyer, R. S. Sundaram, A. Chuvilin, S. Kurasch, M. Burghard, K. Kern and U. Kaiser, Nano Lett., 2010, 10, 1144–1148 CrossRef CAS PubMed.
- T. Sekine, C. Julien, I. Samaras, M. Jouanne and M. Balkanski, Mater. Sci. Eng., B, 1989, 3, 153–158 CrossRef.
- G. Cravotto and P. Cintas, Chem. Soc. Rev., 2009, 38, 2684–2697 RSC.
- H. Stewart, M. Golding, L. Matia-Merino, R. Archer and C. Davies, Colloids Surf., A, 2016, 498, 194–205 CrossRef CAS.
- S. Bari, A. Chatterjee and S. Mishra, Ultrason. Sonochem., 2016, 31, 39–50 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- W. W. Lei, V. N. Mochalin, D. Liu, S. Qin, Y. Gogotsi and Y. Chen, Nat. Commun., 2015, 6, 8849 CrossRef CAS PubMed.
- L. Kihlborg, G. Hägerström and A. Rönnquist, Acta Chem. Scand., 1961, 5, 1187–1188 CrossRef.
- K. A. Wilhelmi, Acta Chem. Scand., 1969, 23, 419–428 CrossRef CAS.
- X. Lai, M. Yan, X. Liu, J. Luo, G. Tang, F. Xie, J. Chen, P. Liu, S. Di, W. Xie and Q. Chen, Mater. Res. Express, 2014, 1, 045044 CrossRef.
- S. Adams, J. Solid State Chem., 2000, 149, 75–87 CrossRef CAS.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- Y. P. Song, H. Wang, Z. H. Li, N. Q. Ye, L. J. Wang and Y. Liu, Int. J. Hydrogen Energy, 2015, 40, 3613–3623 CrossRef CAS.
- L. Huang, X. Gao, Q. Dong, Z. M. Hu, X. Xiao, T. Q. Li, Y. L. Cheng, B. Yao, J. Wan, D. Ding, Z. Ling, J. S. Qiu and J. Zhou, J. Mater. Chem. A, 2015, 3, 17217–17223 CAS.
- W. Xie, M. Su, Z. Zheng, Y. Wang, L. Gong, F. Xie, W. Zhang, Z. Luo, J. Luo, P. Liu, N. Xu, S. Deng, H. Chen and J. Chen, ACS Nano, 2016, 10, 1662–1670 CrossRef CAS PubMed.
- M. Doi and S. F. Edwards, The theory of polymer dynamics, Oxford University Press, 1988 Search PubMed.
- X. D. Zhang, X. Xie, H. Wang, J. J. Zhang, B. C. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18–21 CrossRef CAS PubMed.
- X. D. Zhang, H. X. Wang, H. Wang, Q. Zhang, J. F. Xie, Y. P. Tian, J. Wang and Y. Xie, Adv. Mater., 2014, 26, 4438–4443 CrossRef CAS PubMed.
- Q. Y. Lin, L. Li, S. J. Liang, M. H. Liu, J. H. Bi and L. Wu, Appl. Catal., B, 2015, 163, 135–142 CrossRef CAS.
- M. Wei, Y. Konishi, H. Zhou, H. Sugihara and H. Arakawa, Solid State Commun., 2005, 133, 493–497 CrossRef CAS.
- M. Wei, Y. Konishi, H. Zhou, H. Sugihara and H. Arakawa, Chem. Phys. Lett., 2004, 400, 231–234 CrossRef CAS.
- H. Zhang, ACS Nano, 2015, 9, 9451–9469 CrossRef CAS PubMed.
- J. Xu, S. L. Gai, F. He, N. Niu, P. Gao, Y. J. Chen and P. P. Yang, Dalton Trans., 2014, 43, 11667–11675 RSC.
- C. Suryanarayana and M. G. Norton, X-ray diffraction: a practical approach, Springer Science & Business Media, 2013 Search PubMed.
- J. Y. Luo, L. J. Cote, V. C. Tung, A. T. L. Tan, P. E. Goins, J. S. Wu and J. X. Huang, J. Am. Chem. Soc., 2010, 132, 17667–17669 CrossRef CAS PubMed.
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H. Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568–571 CrossRef CAS PubMed.
- I. Navas, R. Vinodkumar and V. P. M. Pillai, Appl. Phys. A: Mater. Sci. Process., 2011, 103, 373–380 CrossRef CAS.
- L. X. Song, J. Xia, Z. Dang, J. Yang, L. B. Wang and J. Chen, CrystEngComm, 2012, 14, 2675–2682 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra20159h |
| This journal is © The Royal Society of Chemistry 2016 |