
Activity-based protein profiling: an efficient approach to study serine hydrolases and their inhibitors in mammals and microbes
Abstract
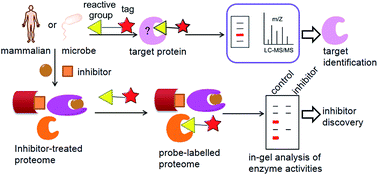
Serine hydrolases (SHs) are involved in a wide range of physiological and pathological processes. Conventional proteomic or genomic methods can only provide an indirect assessment of the functional state of this enzyme in cells and tissues. The lack of effective small-molecule probes or pharmacological tools for the functional characterization of SHs has hindered our understanding of this class of enzymes and its myriad functions and regulation modes. Activity-based protein profiling (ABPP) has emerged as a powerful chemical proteomic method for broad profiling of functional states of enzymes in native biological systems. Herein, we will describe how ABPP has been used to identify and characterize SHs and their inhibitors that are important in physiological and pathological processes of mammals and microbes. Moreover, an integrated workflow for functional mapping of SHs is also depicted.
 Please wait while we load your content...
Please wait while we load your content...

