
Covalent immobilization of glucose oxidase on amino MOFs via post-synthetic modification†
Abstract
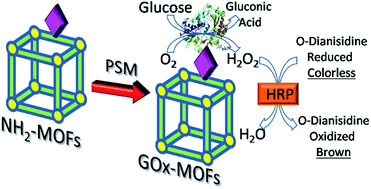
The post-synthetic modification (PSM) of two amino-MOFs with glucose oxidase is reported in this study. The multi-step approach preserved the MOFs' structure and allowed the production of enzyme-functionalized MOFs (MOFs@GOx), which retained the enzymatic activity and showed selective properties for glucose.
 Please wait while we load your content...
Please wait while we load your content...

