DOI:
10.1039/C6RA24042A
(Paper)
RSC Adv., 2016,
6, 108045-108050
Triblock and pentablock copolymerizations of ε-caprolactone with L-lactide catalyzed by N-heterocyclic carbene†
Received
27th September 2016
, Accepted 4th November 2016
First published on 4th November 2016
Abstract
Dihydroxyl capped biodegradable poly(ε-caprolactone)–poly(ethylene glycol)–poly(ε-caprolactone) (PCL–PEG–PCL) triblock copolymer and poly(L-lactide)–poly(ε-caprolactone)–poly(ethylene glycol)–poly(ε-caprolactone)–poly(L-lactide) (PLLA–PCL–PEG–PCL–PLLA) pentablock copolymer have been synthesized by using benzo-12-crown-4-imidazole carbene (B-12-C-4imY) as a catalyst, and PEG and PCL–PEG–PCL copolymer as macroinitiators, respectively. We focused on the influence of the varied reaction time, temperature and monomer/catalyst molar ratio on the copolymerizations. Chemical structures and physical properties of the block copolymers were studied through the characterization of 1H, 13C NMR, GPC and DSC. The NMR spectral data of the copolymers displayed the chemical structures without random placement. Therefore, the pure triblock and pentablock copolymers have been synthesized successfully.
Introduction
Aliphatic polyesters, such as poly(L-lactide) (PLLA), poly(ε-caprolactone) (PCL), poly(glycolide) (PGA) and their copolymers have attracted much attention in various fields, including tissue engineering, bone fixation, and controlled drug delivery due to their bioresorbable and biocompatible properties.1–5 It was reported that PLLA has good physical properties (high strength, thermoplasticity and fabricability). However, hydrophobicity and poor elasticity have limited the applications of PLLA.6 PCL holds high flexibility but degrades very slowly due to its high hydrophobicity and crystallinity, and its mechanical strength is relatively low. Fortunately, it is necessary to adjust the balance of polymer hydrophilicity–hydrophobicity to overcome above problems. Presently, PEG has been widely investigated as a hydrophilic material. When this telechelic PEG is employed as a macroinitiator in the polymerization of hydrophobic monomers, new bifunctional block copolymers with tailored applications can be produced.7
Growing interests have been devoted to the search of efficient catalysts for the copolymerization of ε-CL, LLA and PEG, and different metal systems have been tested and reported, ranging from the traditional stannous octanoate,8 to aluminium compounds,9–12 to zinc catalysts,13 to rare earth complexes.14 However, the cytotoxicity and difficulty in removal of the catalysts from the obtained polymers have limited their use in many cases.15 Purposely, another pathway to prepare polyesters has been developed twenty years ago and consists in the metal-free ring-opening polymerization (ROP) technique, also called organic catalysis. Organic catalysts have been employed for the polymerization of a wide range of monomer types.16 Among them, N-heterocyclic carbenes (NHCs) have emerged not only as versatile ligands for transition metals but also as powerful organocatalysts in molecular chemistry.17 The enormous catalytic potential of NHCs for a large number of polymerization reactions is due to their structural diversity and their varied chemical composition that compete with the most active and selective metal-based or enzymatic catalysts.18 The previous studies have shown that the NHCs are effective for ROP and in the presence of an alcohol initiator generated polymers with high end-group fidelity. Reaction rates can be extremely high and the ROP reaction is remarkably well-controlled and exhibits many of the features of a living polymerization.19 However, to the best of our knowledge, the polymerization with the NHCs modified by the aliphatic and aromatic substituent did not offered high monomer conversion when the polymerization is in progress deviating from the optimal condition.20
In this article, we reported on the synthesis of novel B-12-C-4imY and its application in the ring-opening copolymerization of ε-CL and LLA. The multiblock copolymers of triblock PCL–PEG–PCL and pentablock PLLA–PCL–PEG–PCL–PLLA were synthesized by using dihydroxyl ended PEG and triblock copolymer as macroinitiators, respectively, in the presence of B-12-C-4imY. It was discovered that the high yield of polymerization was obtained (>80%) with variety polymerization reaction condition. The reaction mechanism relating to the high reaction yield is being explored. These copolymers were characterized using 1H, 13C NMR, GPC, DSC and IR.
Experimental
Materials
LLA was prepared from L-lactic acid as described previously21 and recrystallized three times from dried ethyl acetate, followed by drying to constant weight at 40 °C under vacuum. ε-CL (Alfa Aesar) was purified with CaH2 by vacuum distillation in a nitrogen atmosphere. PEG (Mn = 2000) was dried by an azeotropic distillation with dry toluene. Tetrahydrofuran (THF) was dried by refluxing over the blue benzophenone–sodium complex and distilled prior to use. Benzyl alcohol was dried over CaH2 for 2 days, then distilled under reduced pressure and kept over activated 4 Å molecular sieves.
Characterization
Nuclear magnetic resonance (NMR) spectra of the polymers in deuterated chloroform solutions were recorded by a Bruker AV-600 MHz spectrometer with tetramethylsilane as the internal reference. Differential scanning calorimetry (DSC) measurements were carried out on a differential scanning calorimeter (NETZCH DSC 200F3) at a heating rate of 10 °C min−1. The thermal behaviors of copolymers were characterized in the temperature ranging from −60 to 200 °C. The molecular weight and the polydispersity index were determined by a PL-GPC220 gel permeation chromatograph (GPC), using THF as the mobile phase at a flow rate of 1 mL min−1 at 40 °C. The molecular weight calibration curve of polymers was obtained with narrow molecular weight distribution polystyrene standards.
Triblock copolymerization procedure
The PCL–PEG–PCL triblock copolymer was prepared by ROP of ε-CL using PEG as the initiator and B-12-C-4imY as a catalyst. In a typical procedure, the PEG (0.07 g, 0.035 mmol) and ε-CL (0.8 g, 7.0 mmol) were dissolved in THF ([CL] = 2.5 mol L−1). Then, B-12-C-4imY (0.020 g, 0.023 mmol) was added to the reaction mixtures. After the desired reaction time, the product was precipitated by methanol and dried under vacuum to constant weight.
Pentablock copolymerization procedure
As a typical pentablock copolymerization, a 20 mL ampoule was flame-dried under vacuum and charged with ε-CL (0.70 g, 6.13 mmol), the initiator (PEG, 0.0061 g, 0.03 mmol), THF (1.75 mL) and B-12-C-4imY (0.017 g, 0.020 mmol). After 30 min polymerization at 20 °C, LLA (0.88 g, 6.13 mmol) was injected as a THF solution, and the reaction was performed at the desired reaction time. The polymer was precipitated by methanol and dried under vacuum to constant weight.
Results and discussion
Triblock copolymer synthesis
The influences of monomer/PEG ([CL]/[PEG]) and monomer/catalyst ([CL]/[C]) on copolymerization were examined with B-12-C-4imY in THF. Representative results with different [CL]/[PEG] ratio and [CL]/[C] ratio are listed in Table 1. It was found that the excellent [CL]/[PEG] ratio was 200 in the studied range. The yield of the copolymer gradually decreased with increasing [CL]/[PEG] ratio (>200, Table 1, entries 3 and 4). Yet decreasing [CL]/[PEG] ratio (<200, Table 1, entries 1) was found to form more and shorter polymeric chains, thus decreasing the Mn of PCL–PEG–PCL. With fixed [CL]/[PEG] ratio of 200, copolymer molecular weight increased from 35.4 kg mol−1 to 38.0 kg mol−1 as [CL]/[C] molar ratio raised from 250 to 300 (Table 1, entries 2 and 5). Further increasing of [CL]/[C] from 300 to 400 (Table 1, entries 2, 6 and 7) led to a decrease in molecular weight from 38.0 kg mol−1 to 28.4 kg mol−1, because less active species in the reaction medium decreased reaction rate of polymerization.
Table 1 Effects of the monomer/catalyst molar ratio and monomer/PEG molar ratio on the synthesis of PCL–PEG–PCLa
| Entry |
[CL]/[C] |
[CL]/[PEG] |
Yieldb (%) |
Mnc (kg mol−1) |
PDId |
| Condition: [CL] = 2.5 mol L−1, T = 20 °C, t = 30 min. Total reaction yield. Mn determined by GPC. PDI determined by GPC. |
| 1 |
300 |
100 |
92.6 |
22.5 |
1.34 |
| 2 |
300 |
200 |
95.5 |
38.0 |
1.36 |
| 3 |
300 |
300 |
90.1 |
22.4 |
1.52 |
| 4 |
300 |
400 |
88.9 |
22.5 |
1.62 |
| 5 |
250 |
200 |
91.3 |
35.4 |
1.45 |
| 6 |
350 |
200 |
93.7 |
30.8 |
1.36 |
| 7 |
400 |
200 |
88.9 |
23.4 |
1.42 |
The discrepancy of molecular weight and yield with different reaction temperature and time were investigated. As shown in Fig. 1, directly, the triblock copolymerization at 20 °C and 30 min gave the highest yield and molecular weight. The yield and the molecular weight of PCL–PEG–PCL increased with the elevated temperature in the range of 15–20 °C. Higher temperature accelerated the rate of intermolecular transesterification and thermal degradation. Which brought about decreasing molecular weight of PCL–PEG–PCL.22 Time had a great influence on the copolymerization. It was found that the molecular weight of PCL–PEG–PCL increased with increasing of time at 20 °C, until after 30 min the molecular weight of copolymers began to decline and at that point the yield of PCL–PEG–PCL increased to more than 95.5%. Thus the optimum conditions for the triblock copolymerization in THF are [CL]/[PEG] = 200, [CL]/[C] = 300, 20 °C, 30 min.
 |
| | Fig. 1 Effects of reaction temperature and time on the synthesis of PCL–PEG–PCL. aCondition: [M] = 2.5 mol L−1, [M]/[C] = 300, [M]/[PEG] = 200. bTotal reaction yield. cMn determined by GPC. | |
Pentablock copolymer synthesis
Pentablock PLLA–PCL–PEG–PCL–PLLA polymer was prepared by a “one-pot” procedure in the presence of B-12-C-4imY as catalyst without any precursor separation or purification, as shown in Scheme 1. The time and temperature dependence of the copolymerization yield and molecular weight of PLLA–PCL–PEG–PCL–PLLA are listed in Table 2. From the results, the yield of 90.2% and high molecular weight of PLLA–PCL–PEG–PCL–PLLA (Mn = 64.1 kg mol−1) were obtained normally within 60 min at 25 °C. Increasing reaction temperature (>25 °C, Table 2, entries 4 and 5) led to a more rapid copolymerization and copolymer was easily degraded, whereas below the suitable temperature (<25 °C, Table 2, entries 1 and 2) the polymerization reaction proceeded slowly. Prolong reaction time after monomer consumption often broaden the polydispersity, which indicated some transesterification of the polymer backbone occurring.22
 |
| | Scheme 1 Synthesis of PLLA–PCL–PEG–PCL–PLLA pentablock copolymer. | |
Table 2 Effects of reaction temperature and time on the synthesis of PLLA–PCL–PEG–PCL–PLLAa
| Entry |
tc (min) |
Tb (°C) |
Conv.d (%) |
Mne (kg mol−1) |
PDIe |
| Copolymerization conditions: [M] = 2.5 mol L−1, [LLA] = 1.5 mol L−1, [M]/[PEG] = 200. First stage of CL polymerization: 20 °C, 30 min. Polymerization temperature for LLA. Polymerization time for LLA. Total monomer conversion. Mn and PDI determined by GPC. |
| 1 |
60 |
15 |
82.0 |
53.9 |
1.53 |
| 2 |
60 |
20 |
85.9 |
56.1 |
1.35 |
| 3 |
60 |
25 |
90.2 |
64.1 |
1.26 |
| 4 |
60 |
30 |
89.8 |
54.3 |
1.30 |
| 5 |
60 |
35 |
87.5 |
49.3 |
1.46 |
| 6 |
40 |
25 |
81.3 |
55.8 |
1.21 |
| 7 |
50 |
25 |
85.6 |
57.5 |
1.27 |
| 8 |
70 |
25 |
82.4 |
41.9 |
1.34 |
| 9 |
80 |
25 |
80.7 |
37.8 |
1.56 |
Characterization
The average molecular weight and distribution of PCL–PEG–PCL (A) and PLLA–PCL–PEG–PCL–PLLA (B) obtained from GPC are shown in Fig. 2. It could been seen that two tested samples showed single peak, which suggested the monodisperison of Mn and the absence of any homopolymer of ε-CL, LLA and PEG monomer. These indicated that no transesterification of the polymer backbone and/or backbiting reactions occurred. In addition, the GPC trace of the PLLA–PCL–PEG–PCL–PLLA shifted to higher molecular weights, which confirmed the formation of the pentablock structure.
 |
| | Fig. 2 GPC curves of PCL–PEG–PCL (A, sample 2, Table 1) and PLLA–PCL–PEG–PCL–PLLA (B, sample 3, Table 2). | |
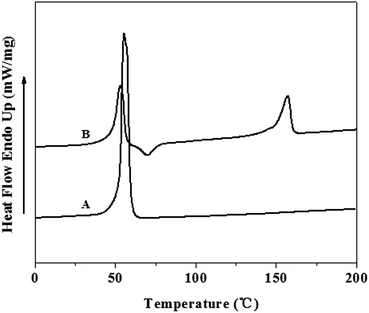
DSC thermograms for PCL–PEG–PCL (A) triblock and PLLA–PCL–PEG–PCL–PLLA (B) pentablock copolymers are depicted in Fig. 3. There was only one endothermic peak (Tm = 55.3 °C) in PCL–PEG–PCL. The phase transition of PEG was not obvious since PCL–PEG–PCL (Table 1, entry 2, Mn = 38.0 kg mol−1) had a long PCL block and short PEG segment. The single phase transition of triblock copolymer has also been reported by others.23,24 Two melting peaks appeared clearly on the thermograms, each melting temperature corresponding to the melting temperature of both homopolymers (53 °C for the PCL segment and 157 °C for the PLLA segment, see Fig. 3(B)).
 |
| | Fig. 3 The DSC curves of PCL–PEG–PCL (A, sample 2, Table 1) and PLLA–PCL–PEG–PCL–PLLA (B, sample 3, Table 2). | |
The presence of PLLA end block attached to triblock prepolymer decreased the melting temperature of corresponding PCL–PEG–PCL block.
Fig. 4 exhibits the FT-IR spectra of the copolymers. Both triblock (A) and pentablock (B) spectra showed an absorption peak at 1105 cm−1 assigned to the characteristic –CH2–O–CH2– stretching vibrations of –OCH2CH2– units from the PEG segment.25 Besides, the spectrum of PCL–PEG–PCL showed that the most important and identifying PCL peak was the ester carbonyl band appearing as a sharp singlet at 1732 cm−1. Compared with PCL–PEG–PCL, in the IR spectrum of the PLLA–PCL–PEG–PCL–PLLA, the carbonyl absorption band became wide and split into two peaks. The peak at 1761 cm−1 belonged to the ester carbonyl group –CH2CO– of PLLA block, while the peak at 1732 cm−1 was the signal of the ester carbonyl group –CH2CO– of the PCL block. These differences were proof of the formation of pentablock copolymer.26,27
 |
| | Fig. 4 IR spectra of PCL–PEG–PCL (A, sample 2, Table 1) and PLLA–PCL–PEG–PCL–PLLA (B, sample 3, Table 2). | |
In order to further confirm the formation of copolymers, the 1H NMR spectra are presented in Fig. 5. Several peaks were observed from both the PCL–PEG–PCL (A) and PLLA–PCL–PEG–PCL–PLLA (B) spectra. Peaks at 1.38, 1.64, 2.30, and 4.05 ppm were assigned to methylene protons of –(CH2)3–, –OCCH2–, and –CH2OOC– in PCL units, respectively, and peak at 3.65 ppm (−CH2CH2–) was assigned to PEG units. What's more, the structure of PEG-PCL diols were confirmed by the peaks at 4.22 ppm belonging to methylene protons of the PCL–CO–OCH2–CH2–O–PEG segment, which indicated that the polymer is triblock (Fig. 5(A)).28 In comparison with the spectrum of PCL–PEG–PCL, new resonances at 5.18 ppm and 1.58 ppm corresponding to the methenyl proton and to the methane proton of PLLA block, respectively, were observed. The peaks at 4.36 ppm were attribute of methenyl proton of lactide end group (Fig. 5(B)). This illustrated that the terminal hydroxyl groups of the PCL–PEG–PCL macroinitiator successfully initiated the polymerization of LLA. The molar ratio of PEG/PCL/PLLA was determined by integrating peak intensities of methylene protons from PEG block at 3.65 ppm, PCL block at 4.05, and to PLLA block at 5.18 ppm. The formation of the pentablock copolymers can be confirmed by the molar ratio of characteristic peaks of each segment.29 The 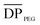 is equal to 45 as derived from the
is equal to 45 as derived from the ![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n, PEG value of 2000 g mol−1. For the triblock,
n, PEG value of 2000 g mol−1. For the triblock, 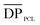 = 260 and n, NMR = 31.7 kg mol−1 (sample 2, Table 1). In addition, for the pentablock,
= 260 and n, NMR = 31.7 kg mol−1 (sample 2, Table 1). In addition, for the pentablock, 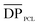 = 207 and
= 207 and  = 370, respectively (sample 3, Table 2). Thus, n, NMR = 52.3 kg mol−1.20,30
= 370, respectively (sample 3, Table 2). Thus, n, NMR = 52.3 kg mol−1.20,30
 |
| | Fig. 5 1H NMR spectra of PCL–PEG–PCL (A, sample 2, Table 1) and PLLA–PCL–PEG–PCL–PLLA (B, sample 3, Table 2). | |
Fig. 6 displays the 13C NMR spectra of PCL–PEG–PCL (B) and PLLA–PCL–PEG–PCL–PLLA (C). For the 13C NMR spectrum of PCL–PEG–PCL, the peak at 173.4 ppm was assigned to the PCL blocks and the peak at 70.4 ppm was attributed to the PEG block (Fig. 6(B)). The 13C NMR spectrum of PLLA–PCL–PEG–PCL–PLLA revealed only two resonances between 160 and 180 ppm, one of the PCL carbonyl (173.7 ppm) and the other of the PLLA carbonyl (169.6 ppm) (Fig. 6(C)). The absence of additional peaks between these two peaks indicated that mixed sequences were not present, as it would be expected if transesterification reactions occurred. Other backbone signals (δ = 70.4, 64.0, 33.9, 28.2, 25.4 and 24.4 ppm) were in close with 13C NMR signal of PCL–PEG–PCL.
 |
| | Fig. 6 13C NMR spectra of PEG (A), PCL–PEG–PCL (B, sample 2, Table 1) and PLLA–PCL–PEG–PCL–PLLA (C, sample 3, Table 2). | |
Possible mechanism
The block copolymerization is through a monomer-activated pathway (Scheme 2).31 First, the ε-CL is activated by the carbene and forms a zwitterionic acylimidazole intermediate. Propagation through chain extension of the PEG species which serve as the nucleophilic alcohol in subsequent propagation affords HO–PEG–PCL. Then, the HO–PEG–PCL is used as a macro-initiator for the ROP of ε-CL to achieve the PCL–PEG–PCL triblock polymer along with the first step. In the meantime, the NHC catalyst is released and can activate another monomer. The reaction is completed when the monomer is consumed. The synthesis of the pentablock is similar.
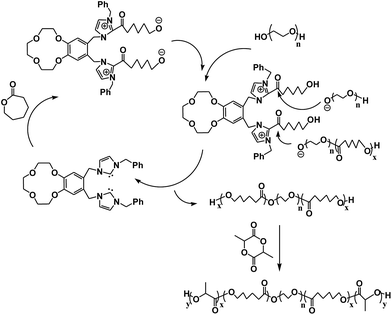 |
| | Scheme 2 Proposed mechanism for the block copolymerization. | |
Conclusions
Here we have reported the effective route to prepare PCL–PEG–PCL triblock and PLLA–PCL–PEG–PCL–PLLA pentablock copolymers using B-12-C-4imY catalyst. PCL–PEG–PCL triblock copolymer with molecular weight of about 38.0 kg mol−1 and molecular weight distribution of 1.36 could be prepared after 30 min at [CL]/[C] = 300 in THF at 20 °C. PLLA–PCL–PEG–PCL–PLLA pentablock copolymer was prepared through a sequential ROP using telechelic PCL–PEG–PCL as macroinitiator. 1H NMR spectroscopy and 13C NMR spectroscopic analyses provided evidence for that there was no transesterification during the block copolymer synthesis. DSC analyses have been performed to assess their thermal properties. Two endothermic peaks were observed for the PLLA–PCL–PEG–PCL–PLLA copolymer, whereas only one endothermic peak was observed for the PCL–PEG–PCL copolymer.
Acknowledgements
This work was supported by Basic Research Project of Shanxi Province of China (No. 2015011029), Undergraduate Innovative Experiment Program of Shanxi Normal University (No. SD2014CXXM-36) and Shanxi Province Education Innovation Project for Postgraduate (No. 2015BY38).
Notes and references
- S. R. Langer, Science, 1993, 260, 920–926 Search PubMed.
- E. Chiellini and R. Solaro, Adv. Mater., 1996, 8, 305–311 CrossRef CAS.
- K. E. Uhrich, S. M. Cannizzaro, S. R. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181–3198 CrossRef CAS PubMed.
- T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 4072–4073 CrossRef CAS.
- B. M. Jeong, Y. H. Bae, D. S. Lee and S. W. Kim, Nature, 1997, 388, 860–862 CrossRef CAS PubMed.
- Y. Hu, Y. S. Hu, V. Topolkaraev, A. Hiltner and E. Baer, Polymer, 2003, 44, 5711–5720 CrossRef CAS.
- E. Jule, Y. Nagasaki and K. Kataoka, Bioconjugate Chem., 2003, 14, 177–186 CrossRef CAS PubMed.
-
(a) D. W. Grijpma, G. J. Zondervan and A. Pennings, Polym. Bull., 1991, 25, 327–333 CrossRef CAS;
(b) Y. Baimark and R. Molloy, ScienceAsia, 2004, 30, 327–334 CrossRef CAS.
-
(a) M. P. F. Pepels, M. Bouyahyi, A. Heise and R. Duchateau, Macromolecules, 2013, 46, 4324–4334 CrossRef CAS;
(b) I. D'Auria, M. Lamberti, M. Mazzeo, S. Milione and C. Pellecchia, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 49–60 CrossRef.
- C. Kan and H. Ma, RSC Adv., 2016, 6, 47402–47409 RSC.
- A. Pilone, N. D. Maio, K. Press, V. Venditto, D. Pappalardo, M. Mazzeo, C. Pellecchia, M. Kol and M. Lamberti, Dalton Trans., 2015, 44, 2157–2165 RSC.
- M. C. Chang, W. Y. Lu, H. Y. Chang, Y. C. Lai, M. Y. Chiang, H. Y. Chen and H. Y. Chen, Inorg. Chem., 2015, 54, 11292–11298 CrossRef CAS PubMed.
- I. D'Auria, M. Lamberti, M. Mazzeo, S. Milione, G. Roviello and C. Pellecchia, Chem.–Eur. J., 2012, 18, 2349–2360 CrossRef PubMed.
- I. D'Auria, M. Mazzeo, D. Pappalardo, M. Lamberti and C. Pellecchia, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 403–413 CrossRef.
- R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS.
- M. Fevre, J. Pinaud, Y. Gnanou, J. Vignolle and D. Taton, Chem. Soc. Rev., 2013, 42, 2142–2172 RSC.
-
(a) D. Enders and T. Balensiefer, Acc. Chem. Res., 2004, 37, 534–541 CrossRef CAS PubMed;
(b) V. Nair, S. Bindu and V. Sreekumar, Angew. Chem., Int. Ed., 2004, 43, 5130–5135 CrossRef CAS PubMed;
(c) N. Marion, S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988–3000 CrossRef CAS PubMed;
(d) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed.
-
(a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed;
(b) N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
-
(a) E. F. Connor, G. W. Nyce, M. Myers, A. Mock and J. L. Hedrick, J. Am. Chem. Soc., 2002, 124, 914–915 CrossRef CAS PubMed;
(b) O. Coulembier, A. P. Dove, R. C. Pratt, A. C. Sentman, D. A. Culkin, L. Mespouille, P. Dubois, R. M. Waymouth and J. L. Hedrick, Angew. Chem., Int. Ed., 2005, 44, 5044–5048 CrossRef;
(c) S. Csihony, D. A. Culkin, A. C. Sentman, A. P. Dove, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2005, 127, 9079–9084 CrossRef CAS PubMed;
(d) G. W. Nyce, T. Glauser, E. F. Connor, A. Mock, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2003, 125, 3046–3056 CrossRef CAS PubMed.
- N. E. Kamber, W. Jeong, S. Gonzalez, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2009, 42, 1634–1639 CrossRef CAS.
- D. K. Giding and A. M. Reed, Polymer, 1979, 20, 1459–1464 CrossRef.
-
(a) Y. Wang, L. F. Zhang, X. N. Guo and R. Zhang, J. Polym. Res., 2013, 20, 87–92 CrossRef;
(b) L. F. Zhang, N. Li, Y. Wang, J. Z. Guo and J. F. Li, Macromol. Res., 2014, 22, 600–605 CrossRef CAS;
(c) Z. X. Zhu, C. D. Xiong, L. L. Zhang and X. M. Deng, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 709–714 CrossRef CAS.
- Y. Zhang and R. X. Zhuo, Biomaterials, 2005, 26, 6736–6742 CrossRef CAS PubMed.
- M. J. Hwang, J. M. Suh, Y. H. Bae, S. W. Kim and B. Jeong, Biomacromolecules, 2005, 6, 885–890 CrossRef CAS PubMed.
- C. B. Liu, C. Y. Gong, M. J. Huang, J. W. Wang, Y. F. Pan, Y. D. Zhang, G. Z. Li, M. L. Gou, K. Wang and M. J. Tu, J. Biomed. Mater. Res., Part B, 2008, 84, 165–175 CrossRef PubMed.
- Q. Cai, J. Z. Bei and S. G. Wang, Polym. Adv. Technol., 2000, 11, 159–166 CrossRef CAS.
- W. Z. Yuan, X. Z. Tang, X. B. Huang and S. X. Zheng, Polymer, 2005, 46, 1701–1707 CrossRef CAS.
-
(a) G. Z. Yin, D. L. Zhao, X. Wang, Y. Ren, L. W. Zhang, X. X. Wu, S. P. Nie and Q. F. Li, RSC Adv., 2015, 5, 79070–79080 RSC;
(b) Z. Draoua, A. Harrane and M. Belbachir, J. Macromol. Sci., Part A: Pure Appl.Chem., 2015, 52, 130–137 CrossRef CAS.
- V. Tamboli, G. P. Mishra and A. K. Mitra, Colloid Polym. Sci., 2013, 291, 1235–1245 CAS.
-
(a) H. Nouailhas, F. Li, A. E. Ghzaoui, S. Li and J. Coudane, Polym. Int., 2010, 59, 10771083 Search PubMed;
(b) A. Harrane, A. Leroy, H. Nouailhas, X. Garric, J. Coudane and B. Nottelet, Biomed. Mater., 2011, 6, 065006 CrossRef PubMed.
- J. Raynaud, Y. Gnanou and D. Taton, Macromolecules, 2009, 42, 5996–6005 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Catalyst preparation process. See DOI: 10.1039/c6ra24042a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 





 is equal to 45 as derived from the
is equal to 45 as derived from the  = 260 and
= 260 and  = 207 and
= 207 and  = 370, respectively (sample 3, Table 2). Thus,
= 370, respectively (sample 3, Table 2). Thus, 

