
Enzymeless biosensor based on β-NiS@rGO/Au nanocomposites for simultaneous detection of ascorbic acid, epinephrine and uric acid†
P. Muthukumarana,
C. Sumathia,
J. Wilson*a and
G. Ravib
aPolymer Electronics Lab, Department of Bioelectronics and Biosensors, Alagappa University, Karaikudi-630004, Tamilnadu, India. E-mail: wilson.j2008@yahoo.com
bPhotonic Crystals Lab, Department of Physics, Alagappa University, Karaikudi-630 004, Tamilnadu, India
First published on 28th September 2016
Abstract
In this study, marigold flower-like self-assembled β-NiS (nickel sulfide) nanosheets were grown on rGO (reduced graphene oxide) by a single-step hydrothermal process and then gold nanospheres (AuNS) were electrochemically deposited on the β-NiS@rGO nanostructures. The β-NiS@rGO/AuNS hierarchical hybrid demonstrates excellent electrochemical activities for clinically important analytes such as AA (ascorbic acid), EP (epinephrine) and UA (uric acid) simultaneously. Moreover, the β-NiS@rGO/AuNS/GCE exhibits significant detection of AA by one order and UA by two orders higher sensitivity when compared to a β-NiS@rGO modified electrode. We investigated using this electrocatalyst for individual detection, the linear responses of AA, EP and UA at concentrations in the ranges of 900 nM to 100 μM, 2 μM to 1 mM and 100 nM to 1 mM with low detection limits of 331 nM, 540 nM and 4.5 nM (S/N = 3σ/b), respectively; the resulting non-enzymatic biosensor also retained a good response to simultaneous detection with a wide linear range 1 μM to 1 mM, 2 μM to 1 mM and 100 nM to 1 mM and significantly lower detection limits of 682 nM, 1.3 μM and 6 nM, respectively. Excellent stability, reproducibility, and selectivity were achieved. Biosensing of AA, EP and UA in human urine, blood, serum, fruits and pharmaceutical drugs was successfully demonstrated without any preliminary treatment.
1. Introduction
The control and management of clinical analytes like ascorbic acid (AA), epinephrine (EP) and uric acid (UA) in the extracellular fluids plays a magnificent role in the organic function of the human body's metabolism. EP, a hormone secreted by medulla of the adrenal glands, is a significant catecholamine neurotransmitter in the health of the central nervous system of humans and other mammals. EP has been known to be related to various neurological diseases such as Parkinson's disease, whereas AA and UA have been coupled to gout and hyperuricemia. Therefore the control of these compounds is of great importance. It is of colossal need to develop an accurate simultaneous voltammetric measurement of these analytes in finding solution for health care problems.1–5 The clinically required detection range of AA for well-nourished, non-smoking individuals is 50–60 μM; the epinephrine range is 54.59–545 nM and the uric acid (serum) range is 180–420 μM.Hence, it is pertinent to investigate and design highly sensitive biosensors with low detection limits to achieve convincing clinical measurements. Therefore, it is very important to fabricate cheaper and simpler devices by eliminating the challenges in the conventional electrodes, such as fouling of electrode surfaces and overlap at their oxidation potentials and surface poisoning from adsorbed intermediates. Several approaches based on different modified electrodes including conducting polymers,6 dendrimers,7 CPE/MWCNT,8 rGO–CNT/ITO,9 Pd–Au/GCE10 and LaMnO3/GCE11 have been developed in the past for electrochemical simultaneous detection of DA, AA and UA. However, these approaches involving environmentally unfriendly organic solvents during the preparation process, aggravated interference due to other electroactive analytes and poor electrochemical conductivity limit their applications.
In contrast, transition metal sulfides are a promising class of materials in terms of their high reversible capacity, long cycle stability and intrinsic safety features.12–15 Based on their electrochemical performances, a few metal sulfides attracted several researchers due to their superior electrical conductivity in biosensor applications.16–18 However, they suffer from insufficient sensitivity to achieve low-level detection, poor ion transport and less mechanical stability. To circumvent these inconveniences, in this decade numerous efforts have been made for the development of hybrid nanostructures through incorporating graphene materials19–22 (of greater conductivity, flexibility, high chemical stability, and large surface area). Decoration of nanostructured metal sulfides on graphene honeycomb lattice is anticipated to relieve aggregation of active materials and enhance the conductivity. These metal sulfide–graphene-based composites (CoS–graphene, NiS–graphene and CuS–graphene) effectively enhanced the electrocatalytic properties and reduced the aggregation of active materials.23–25
Among the noble metal-free catalysts, nickel sulfide (β-NiS) is not only inexpensive but could also replace the others because of its versatile applications in super capacitors,26 hydrogenation catalysts,27 dye-sensitized solar cells28 and lithium ion batteries.29 However, significantly improving the electrochemical activity and designing low-cost and robust biosensors remains challenging.
Singh et al. revealed that the MWCNT incorporated with NiS could enhance the electrochemical property to exhibit 1000 cycles that retained 92% of asymmetric super capacitor behavior.30 Wei et al. reported that a mesoporous ZnS–NiS nanocomposite possessed efficient glucose sensing ability with a detection limit of 0.125 μM.31 Similarly, Ni0.31Co0.69S2/rGO and NiS/rGO catalysts demonstrated the high performance non-enzymatic glucose sensor of linear range 0.001–5 mM with the lowest detection limit of 0.078 μM and 5.0 × 10−5 to 1.7 × 10−3 M with detection limit of 1.0 × 10−5 M, respectively.32,33
Recently the exfoliation method has attracted tremendous interest in the electrocatalytic reactions because of its remarkable chemical, physical, conductivity and surface area behavior. Novoselov et al. applied a new exfoliation method resulting in high yield and large scale production, in which reduced graphene oxide (rGO) was used for the conversion of NiS nanorods into NiS nanosheets.34 The presence of the oxygen groups in rGO makes it possible to functionalize with NiS through covalent and non covalent methods.35 In addition, strong hybridization between NiS and rGO could also be established through possible chemical bonding.36 A large fraction of studies has been focused on exploring new functional applications of gold nanospheres (AuNS) in electrochemical biosensors based on their catalytic activity.37,47 Interestingly, the interaction of NiS with AuNS effectively enhanced the photocatalytic activity for the hydrogen evolution reaction in the AuNS@NiS@CNT hybrid nanocatalyst.38
Inspired by this, we report the fabrication and characterization of an electrochemical biosensor based on rGO-embedded β-NiS nanosheets with functionalized AuNS to detect, simultaneously, AA, EP, and UA. To gain insight into the intrinsic transport properties and performance of AuNS decorated on self-assembled β-NiS@rGO hybrid nanostructure, we thoroughly characterize the sample using scanning electron microscopy (SEM), X-ray diffraction (XRD), energy-dispersive X-ray spectroscopy (EDS), ultraviolet-visible (UV-Vis) spectroscopy, Raman spectroscopy and on an electrochemical workstation. Thus, the β-NiS@rGO/AuNS nano hybrid, in which a strong interaction could be realized through chemical bonding, is a promising substitute for precious materials in biosensor device fabrication (Scheme 1).
 | ||
| Scheme 1 Schematic illustration of simultaneous detection of AA, EP and UA using AuNS decorated on self-assembled β-NiS@rGO nanocomposite. | ||
2. Experimental
2.1. Materials and reagents
Potassium nitrate, gold chloride (HAuCl4·3H2O), graphite flakes, nickel sulfate heptahydrate, thiourea, sodium nitrate, potassium permanganate, hydrogen peroxide, ethanol, and ammonia were obtained from Sigma Aldrich. AA, UA and EP were purchased from Sisco Research Laboratories. Vitamin C tablets and EP hydrochloride injectable dose was received from Domin, Neon Laboratory Ltd. 0.1 M phosphate buffer solution (PBS) with different pH values was prepared by mixing with the stock standard solutions of Na2HPO4 and NaH2PO4, and the pH was adjusted with 0.1 M H3PO4 or NaOH. Aqueous solutions used throughout were prepared with ultra-pure water obtained from a Millipore system. AA, EP and UA solutions were freshly prepared in PBS prior to use. All the purchased reagents were used without any further treatments. The fresh human real samples such as blood, serum and urine were obtained from Aravind Biochemical Lab., Karaikudi.2.2. Instruments and measurements
The surface morphology of the samples was investigated by scanning electron microscopy (SEM); X-ray diffraction (XRD) was performed using a Bruker D8 Advance (Germany) with Cu-Kα radiation (1.5418 Å) and the Raman spectrum was obtained using an imaging spectrograph (model STR 500 mm focal length) laser Raman spectrometer (SEKI, Japan). UV-Vis studies were carried out using a Shimadzu UV-1700 spectrophotometer. Electrochemical experiments were performed using a CHI 6005D electrochemical workstation (Austin, USA) using a GC working electrode (0.07 cm−2), an Ag/AgCl (3.0 M KCl) reference electrode and a platinum wire auxiliary electrode. All the measurements were carried out in PBS as a supporting electrolyte under a nitrogen atmosphere at room temperature. The origin 8 software was used to calculate the surface area of the modified electrodes. Prior to surface modification, the bare GCE (glassy carbon electrode) was polished with gamma alumina suspensions of 1.0, 0.3 and 0.05 mm. Furthermore, the electrode was successively washed in ethanol and DD (doubly-distilled) water for 15 minutes by an ultrasonic method. The GCE was modified with the β-NiS@rGO/AuNS ternary nanostructured sample and dried at room temperature without any pretreatment of the electrode surface. Finally, different concentrations of AA, EP and UA were injected into the 0.1 M PBS separately and simultaneously, and their corresponding current waves were recorded. In order to verify the reliability of the proposed biosensor, the real sample analysis was performed as follows; for AA analysis, in pharmaceutical products and fruits, the procedure was as below: the tablets were gently pulverized and dissolved in DD water and diluted with PBS (pH 7; 0.1 M) at different concentrations; lemon extract was diluted 10 times in PBS and then AA presence in it was investigated. Furthermore, to evaluate the EP investigation, a commercial epinephrine hydrochloride injection of 100 μL (1 mg mL−1) was diluted in PBS without any pretreatment. Similarly, the urine samples were diluted 10 times with 0.1 M PBS solution (pH 7.0). We also attempted the simultaneous measurements of AA, EP and UA from fresh human blood and serum using 10 mL of 0.1 M PBS. The standard addition method was used to evaluate the content of analytes in all the real samples.2.3. Synthesis of β-NiS@rGO hybrid nanostructure
Graphene oxide (GO) was prepared through the modified Hummers, method.39 Typically, 15 mM of nickel sulfate heptahydrate, dissolved in 80 mL of DD water, was mixed with 30 mM of thiourea and stirred until a clear transparent light-green solution was obtained. To this, 5 mL of ammonia solution was added drop by drop at room temperature under vigorous stirring for 1 h to obtain a color change from a light-green to a blue solution. Afterwards 50 mg of GO dispersed in 10 mL ethanol solution was sonicated for 1 h and added to the above transparent blue solution, and stirring was continued for 1 h. Finally, the black slurry colored solution was transferred into a Teflon-lined autoclave and hydrothermally heated at 160 °C for 24 h. Finally, the autoclave was cooled to room temperature, centrifuged and repeatedly washed with DD water followed by ethanol. The resulting product was dried in a vacuum oven at 60 °C for 8 h and calcined at 400 °C for 2 h in a horizontal tubular furnace under a nitrogen flow atmosphere. For comparison, the same procedure was used for the preparation of β-NiS nanostructures without the addition of GO.2.4. Electrochemical deposition of AuNS on β-NiS@rGO composite
The as-synthesized β-NiS@rGO composite was drop casted on GCE, dried for 1 h at room temperature and thoroughly rinsed with water to fabricate the modified electrode. On the other hand, 51 mM of gold chloride (HAuCl4·3H2O) was dissolved with DD water in a 100 mL beaker to make a stock solution. 0.1 g KNO3 was added to 10 mL of DD water and dissolved completely in a separate beaker. Then 3 mM of well shaked golden yellow stock solution was mixed after replacing 3 mM from the above solution. Subsequently, using the chronoamperometry technique, the AuNS were electrochemically deposited on the β-NiS@rGO composite at a fixed potential −0.1 V for 350 seconds.3. Results and discussion
3.1. Structural analysis
The purity, crystallinity and the phase formation of the prepared products synthesized are determined by XRD as shown in Fig. 1(A). The XRD pattern of wrinkled GO nanosheets distinguishable by two identical peaks appearing at 10.35°, 42.1° associated with (001) and (111) confirms its presence.40 Unfortunately, in the rGO pattern, the characteristic peaks at 10.35° and 42.1° diminished due to oxygen functional group removal when the reduction process occurred; as a result of this, a weak, broader peak arose at 24.2° (for 002).41,42 The β-NiS self-assembled nanosheets with reported 2θ values of 18.3°, 30.2°, 32.1°, 35.6°, 40.4°, 48.7°, 52.6° were indexed as (110), (101), (300), (021), (211), (131), and (401), respectively, as a single phase of rhombohedral millerite β-NiS.43,44 All diffraction peaks in this pattern are in good agreement with the JCPDS card no: #86-2281. There are no nickel oxide or hydroxide characteristic peaks detected, depicting the purity of the material. As presented in the XRD pattern of β-NiS/rGO composite in which the hydrothermal reduction process of GO started to anchor on β-NiS, shows the diffraction peaks of rGO diminished due to hydrophobic characteristics, however the rGO network supports attractive electron transfer in the composite as reported.45 These results were further supported by the SEM characterization and indicated that rGO could assist the growth of NiS because of the confining function of rGO. Small hump peaks of AuNS at 38°, 45°, 65° corresponding to (111), (200), (220) of a face-centered cubic structure were seen in the β-NiS@rGO/AuNS hybrid nanostructure.46,47 | ||
| Fig. 1 (A) XRD patterns (a) GO (b) rGO (c) β-NiS (d) β-NiS@rGO (e) β-NiS@rGO/AuNS (B) UV-visible spectra (a) β-NiS (b) GO (c) β-NiS@rGO (d) β-NiS@rGO/AuNS (C) Raman spectra (a) GO (b) β-NiS (c) β-NiS@rGO (d) β-NiS@rGO/AuNS. | ||
In Fig. 1(B), the absorption spectrum of GO with well π–π* plasmon transition of aromatic bond (C–C) having sharp peak at 230 nm with energy band gap 4.5 eV and a shoulder peak at 300 nm are observed due to π–π* plasmon transition of aromatic (C–C) bond and n–π* transition (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) bond as reported by Goki et al.48 β-NiS intertwined nanosheets are confirmed by a single weak broader peak appearing nearer to 227 nm.49 In the spectrum of β-NiS@rGO composite, the peak obtained at 230 nm is slightly shifted to 270 nm, whereas the shoulder peak at 300 nm disappears suggesting the reduction of GO to rGO. In the AuNS deposited over β-NiS@rGO sample, several weak and broader peaks at 520 and 540 nm are seen and well proven evidence for the existence of AuNS over the β-NiS@rGO composite.3,50
O) bond as reported by Goki et al.48 β-NiS intertwined nanosheets are confirmed by a single weak broader peak appearing nearer to 227 nm.49 In the spectrum of β-NiS@rGO composite, the peak obtained at 230 nm is slightly shifted to 270 nm, whereas the shoulder peak at 300 nm disappears suggesting the reduction of GO to rGO. In the AuNS deposited over β-NiS@rGO sample, several weak and broader peaks at 520 and 540 nm are seen and well proven evidence for the existence of AuNS over the β-NiS@rGO composite.3,50
The Raman spectrum in Fig. 1(C) evidenced the structural information and existence of the samples in the ternary flexible structure. The GO spectrum exhibits two characteristic peaks appearing around the D band (at 1353 cm−1) due to sp3 defects and the G band (at 1597 cm−1) associated with in-plane vibration of sp2 carbon atoms.51 On investigation of β-NiS nanosheets, the peaks around 241, 369, 1152 cm−1 and the D band (1301 cm−1) and G band (1597 cm−1) support its presence. Notably, small water-absorbed molecule peaks namely 1500–1700 cm−1 were seen. Also the peak at 3253 cm−1 could represent the symmetric stretching vibration of β-NiS.43,52 In addition, in the β-NiS@rGO same spectrum, slight hump peaks were seen at 369 and 518 cm−1 due to sulfate ions as a result of adsorption of water molecule during hydrothermal treatment; the D band remained unchanged (1301 cm−1), whereas the G band slightly downshifted to 1587 cm−1, clearly showing the successful reduction process of GO into rGO. The spectrum of β-NiS@rGO/AuNS hybrid shows a small upshifted G band from 1596 to 1601 cm−1, which could be attributed to the deposition of AuNS on β-NiS@rGO nanosheets.50,51,53
3.2. SEM analysis
Fig. 2(A) shows surface morphology of GO wrinkled nanosheets randomly compact and stacked together, showing uniform laminar morphology, whereas the image of rGO in Fig. 2(B) depicts rippled and entangled with each other, resembling silk veil waves.3,54,55 The low magnification image of β-NiS is shown in Fig. 2(C); the marigold flower-like β-NiS nanostructure in Fig. 2(D) is shown at high magnification.56,57 Fig. 2(E) clearly exhibits that the β-NiS nanoparticles are well attached and distributed on the rGO sheets and also it is difficult to distinguish them from each other. The S and Ni elements exhibit continuous and uniform distribution in rGO, indicating that β-NiS sheets are homogeneously dispersed in rGO sheets. As a result, the composite gets sheet-like morphology. This happens due to the following proposed mechanism: (i) initially, NiS crystallizes into rods and (ii) the oxygen-containing groups and defects of rGO can serve as the nucleation sites for NiS sheets, inhibiting them from growing into a rod-like structure. By the process of etching, the β-NiS nanorods are exfoliated/penetrated into rGO sheets.56,58 Fig. 2(F) shows that AuNS have been effectively assembled on the surface of β-NiS@rGO nanosheets.4,59 Moreover, EDS measurements confirm the elements present in the hybrid nanostructure (Fig. S1†). | ||
| Fig. 2 SEM morphology (A) GO (B) rGO (C) & (D) low and high magnification of marigold flower-like β-NiS (E) β-NiS@rGO (F) β-NiS@rGO/AuNS hybrid nanostructure. | ||
3.3. Electrochemical properties of modified GC electrodes
Cyclic voltammetry (CV) performed in the potential range of −0.2 to 0.8 V in the GCE in presence of 1 mM [Fe(CN)6]3−/4− in 0.1 M KCl solution is shown in Fig. 3(A). The cathodic peak current for bare GC is 2.202 × 10−5 A, and on the deposition of self-assembled nanosheet β-NiS on GC, the peak current remarkably decreases to 3.613 × 10−6 A due to electron barrier properties. Similarly, drop casting of GO on GC exhibits irreversible redox behavior on [Fe (CN)6]3−/4− resulting in a decreased current of 1.227 × 10−5 A. Whereas, for β-NiS@rGO/GCE it shows 2.369 × 10−5 A and this could be attributed to the high surface area and good electrical conductivity of the NiS@rGO system which diffuses more [Fe(CN)6]3−/4− redox couple on its surface. The co-electron cloud between β-NiS and rGO sheets could significantly improve the electronic conductivity. The graphene of the β-NiS/rGO nanohybrid could confine the growth of the NiS cocatalyst to expose more active sites and act as an excellent electron conductor to efficiently transfer electrons from the analytes to catalytic active sites of hybrid nanostructure and NiS also restricts the formation of face-to-face stacking in rGO sheets. Moreover, the enhanced high surface area is favorable for more active sites and facilitates the transport of the electrolyte ions and also diffusion and migration paths of electrolyte ions are greatly shortened to enhance the conductivity. Interestingly, AuNS decorated on β-NiS@rGO nanosheet produces maximum current to 1.045 × 10−4 A due to an increase in surface area and surface roughness of AuNS on the β-NiS@rGO surface. We have also investigated a different scan rate (10–100 mV s−1) for β-NiS, GO, β-NiS@rGO and NiS@rGO/AuNS in [Fe (CN)6]3−/4− solution (Fig. S2†). | ||
| Fig. 3 (A) CV of the GCE in presence of 1 mM [Fe(CN)6]3−/4− in 0.1 M KCl at a scan rate of 50 mV s−1 (B) EIS behavior measured by impedance in the frequency range of 100 kHz to 0.1 Hz; (curve a) bare GC, (curve b) pure GO, (curve c) β-NiS, (curve d) β-NiS@rGO, (curve e) β-NiS@rGO/AuNS modified electrodes. | ||
Similarly, impedance spectroscopy was used to show the electron transfer capability of different modified electrodes investigated by electrochemical impedance analysis and the corresponding results are shown in Fig. 3(B). The charge transfer resistance (Rct) values were obtained by the Randels equivalent circuit model [R(Q)R(O)] using Zsimpwin software. The Rct at different stages was altered with the successive modified electrodes with [Fe(CN)6]3−/4− as the redox probe. The Rct values of bare, β-NiS, GO, β-NiS@rGO, β-NiS@rGO/AuNS/GCE were 638, 4239, 3832, 116, 62 Ω cm−2, respectively. It is clear that the Rct value of the β-NiS@rGO/AuNS sample shows a low value indicating more affinity towards the electrochemical reaction at the electrode/electrolyte interface.
3.4. Determination of electrochemical oxidation of AA, EP and UA
In Fig. 4(A) there are no distinguishable peaks observed for the bare, β-NiS- and GO-modified electrodes in the presence of 1 mM AA, EP and UA in 0.1 M PBS (pH 7.0). Interestingly, on modification with β-NiS@rGO/GCE (curve d), the oxidation peaks are noted at the potentials 0.18, 0.15, and 0.33 V for the analytes with currents of 1.9 × 10−7 A, 2.9 × 10−5 A and 2.3 × 10−5 A, respectively. This is due to the fact that this layered nanoarchitecture facilitates the electrolyte access, as well as enlarges the contact area between the nanocomposite and PBS for improved transport kinetics. The attachment of AuNS to the β-NiS@rGO/GCE resulted in potential shift with remarkable increased current of 7.4 × 10−6 A, 6.5 × 10−5 A and 5.4 × 10−5 A for AA, EP and UA, respectively (curve e). This indicates that AuNS efficiently catalyzed the electrochemical oxidation of analytes. Fig. 4(B) shows the effect of scan rate on oxidation at the β-NiS@rGO/AuNS/GCE in 0.1 M PBS containing 1 mM of AA, EP and UA. It can be seen that the anodic peak shifts slightly to the negative side and the cathodic peak shifts towards the positive side with the increase in scan rate (10–500 mV). These results indicate that the redox reaction of PBS on the β-NiS@rGO/AuNS hybrid nanostructure is rapid and reversible. Moreover, the cathodic peak potential shifts positively, suggesting that there is a kinetic limitation in the reaction of AA, EP and UA oxidation.60 A linear relationship of ipc with scan rate1/2 is observed (Fig. 4(B)), suggesting that AA, EP and UA are electrocatalytically oxidized on the surface of the β-NiS@rGO/AuNS/GCE hybrid through diffusion-controlled electrode processes. Even though the plots of ipc vs. scan rate1/2 exhibit similar slopes for the AA, EP and UA at the β-NiS@rGO/AuNS/GCE, the slope for EP is steeper than that of other analytes, which verifies the best electrocatalytic behavior among the 3 biomolecules simultaneously investigated (Fig. 4(C)). The individual response of AA, EP and UA (500 μM) on β-NiS@rGO/AuNS/GCE is also investigated (Fig. S3†). | ||
| Fig. 4 (A) CV behavior curve (a) bare GC (b) β-NiS (c) GO (d) β-NiS@rGO (e) β-NiS@rGO/AuNS. (B) β-NiS@rGO/AuNS modified GC electrode for different scan rates 10–500 mV s−1. (C) Effect of scan rate at β-NiS@rGO/AuNS modified GCE in presence of 1 mM AA, EP and UA in 0.1 M PBS (pH 7.0). (D) Effect on pH from 1 to 9 at β-NiS@rGO/AuNS modified GCE in presence of 600 μM AA, EP and UA. | ||
The performance of pH value for electrocatalytic response for β-NiS@rGO/AuNS/GCE towards the 600 μM of AA, EP and UA at 50 mV s−1 is seen in Fig. 4(D). Unidentified peaks were observed (pH 1 to 4) in both anodic and cathodic potentials due to electrostatic repulsive force between cationic analytes and the surface of the electrode because of the hefty amount of positive ion accumulation on the β-NiS@rGO/AuNS/GCE surface in the acidic conditions, whereas a slight increase in peak intensities was clearly visible at pH 5 and 6. Interestingly, at pH 7, the amounts of negative ions on the electrode surface were found rather than positive ions to benefit the electrostatic attractive force resulting in sharp peaks. However, at pH 8 the peaks diminished suddenly, whereas at pH 9 broader peaks were noticed. These results clearly demonstrate that the redox reaction undergoes an electron/proton process and pH 7 is the optimal condition for further investigation.11
3.5. Individual and simultaneous determination of AA, EP, and UA
On β-NiS@rGO/GCE, the SWV oxidation peak potentials of AA, EP and UA were at −0.33, 0.27, and 0.34 V, respectively, and the oxidation peak currents of these analytes increased linearly with increasing concentration. The linear responses for the individual determinations of AA, EP and UA are observed in the linear concentration ranges of 10 μM to 1 mM, 10 μM to 1 mM, and 10 μM to 1 mM (Fig. S5(A–C)†), respectively. This is believed to be due to the transformation of rGO electrons to the suitable Ni 3d and S 3p hybrid orbitals and could facilitate the oxidation process of the analytes. Even though β-NiS nanorods on rGO self-assembled nanosheets favor homogenous dispersion and the possibility for hydrophilicity due to the electrostatic attraction between positively charged Ni2+ and negatively charged graphene oxide sheets, the sensitivity is not sufficient for the individual and simultaneous wide-range detection of AA, EP and UA. On the other hand, in β-NiS@rGO/AuNS/GCE, the SWV oxidation peak potentials of AA, EP and UA were at 0.11, 0.14, and 0.29 V, with individual detection in the ranges 900 nM to 100 μM, 2 μM to 1 mM and 100 nM to 1 mM and limits of detection of 331 nM, 540 nM, and 4.5 nM (S/N = 3σ/b), respectively (Fig. 5–7). On comparison of the above two results for individual detection, it is observed that the AuNS-modified electrode shows one order for AA and two orders for UA higher sensitivity.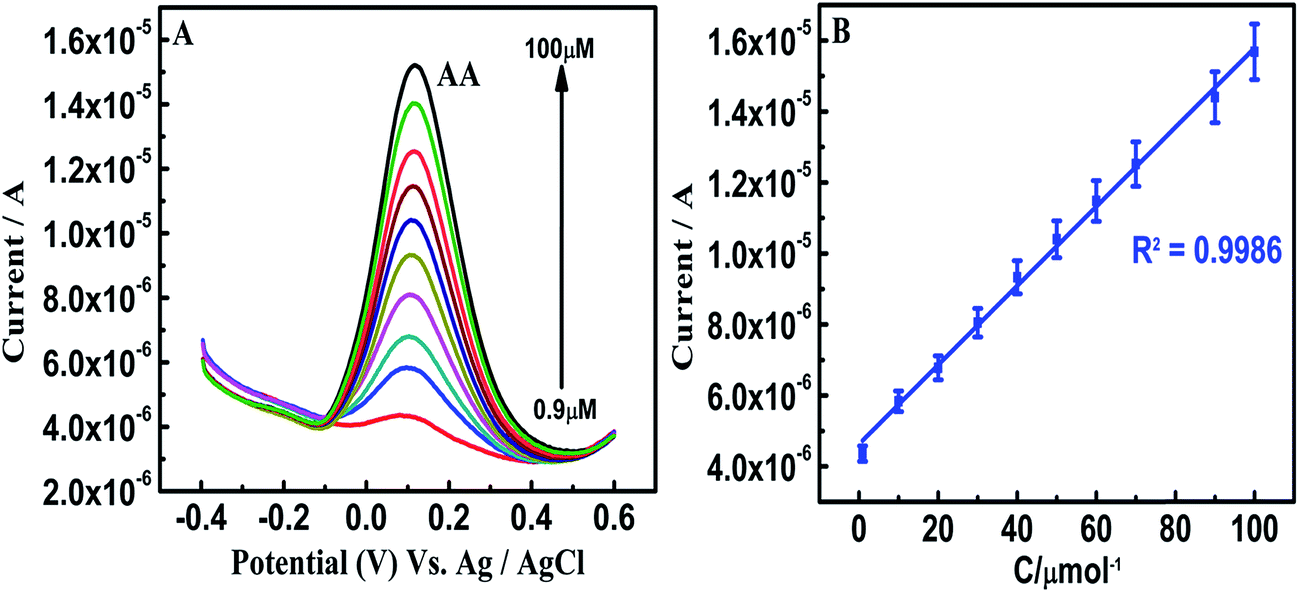 | ||
| Fig. 5 (A) SWV curves 0.9–100 μM AA on β-NiS@rGO/AuNS/GCE in 0.1 M PBS (pH 7.0) (B) calibration plot of the oxidation peak current against different concentrations of AA. The error bars represent standard deviations for five tests. | ||
 | ||
| Fig. 6 (A) SWV curves 2 μM to 1 mM EP on β-NiS@rGO/AuNS/GCE in 0.1 M PBS (pH 7.0) (B) calibration plot of the oxidation peak current against the different concentrations of EP. The error bars represent standard deviations for five tests. | ||
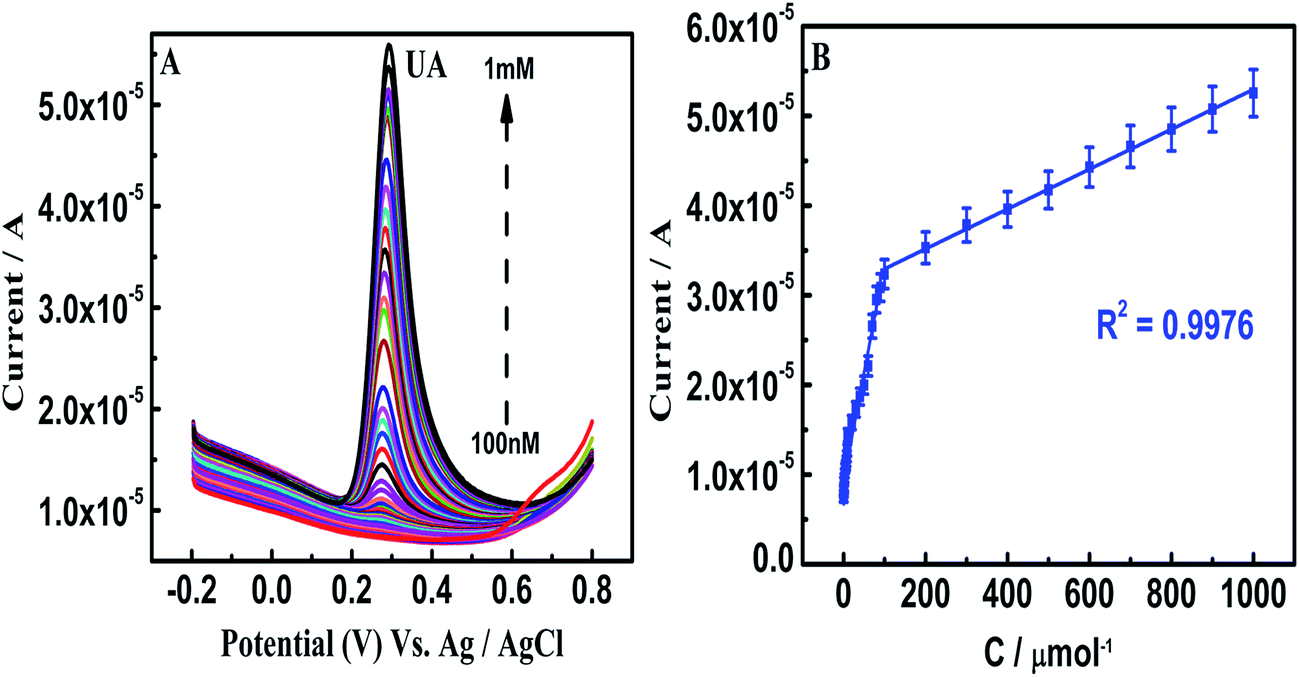 | ||
| Fig. 7 (A) SWV curves 100 nM to 1 mM UA on β-NiS@rGO/AuNS/GCE in 0.1 M PBS (pH 7.0) (B) calibration plot of the oxidation peak current against different concentrations of UA. The error bars represent standard deviations for five tests. | ||
The superior electrochemical properties of AuNS in the hybrid structure could be well explained by the following aspects:
(i) The van der Waals' binding between AuNS and rGO is strong enough to retain AuNS on the surface of the hybrid nanostructure.61
(ii) AuNS can form strong and localized plasmonic near fields to enhance the electron–hole pair generation.62
(iii) The electrostatic interaction between β-NiS and AuNS is also believed to enhance the redox process.63
(iv) The surface area of the modified β-NiS@rGO/AuNS/GCE for AA (4.36 × 10−6 cm−2), EP (7.54 × 10−6 cm−2), and UA (2.85 × 10−6 cm−2) is one order higher compared to β-NiS@rGO composite for AA (8.13 × 10−7 cm−2), EP (5.35 × 10−7 cm−2), and UA (5.81 × 10−7 cm−2), respectively. Hence, this greater surface area in the AuNS modified electrode enhances the catalytic behavior.
(v) AuNS has reduced the oxidation potential of individual detections of AA, EP and UA by 0.22, 0.13, and 0.05 V respectively.
Furthermore, the β-NiS@rGO/AuNS/GCE is used to determine one analyte when the other two analytes are present at fixed concentrations (Fig. 8–10). The SWV oxidation peak current of AA increased linearly with its concentration in the range of 30–800 μM, whereas those of EP (50 μM) and UA (50 μM) remaining almost unchanged. No evident interference is observed for the determination of AA in the presence of EP and UA. Similar cases occurred for the determination of EP in the concentration range 10 μM to 1 mM, whereas the concentrations of AA (at 90 μM) and UA (at 40 μM) were kept constant; UA was found in the concentration range 40 μM to 1 mM, whereas the concentrations of AA (at 90 μM) and EP (at 10 μM) were kept constant.
 | ||
| Fig. 8 (A) SWV of different concentrations of AA 30–800 μM in the presence of fixed concentrations EP and UA on β-NiS@rGO/AuNS/GCE in 0.1 M PBS (pH 7.0) (B) calibration plot for different concentrations of AA in the presence of EP and UA. | ||
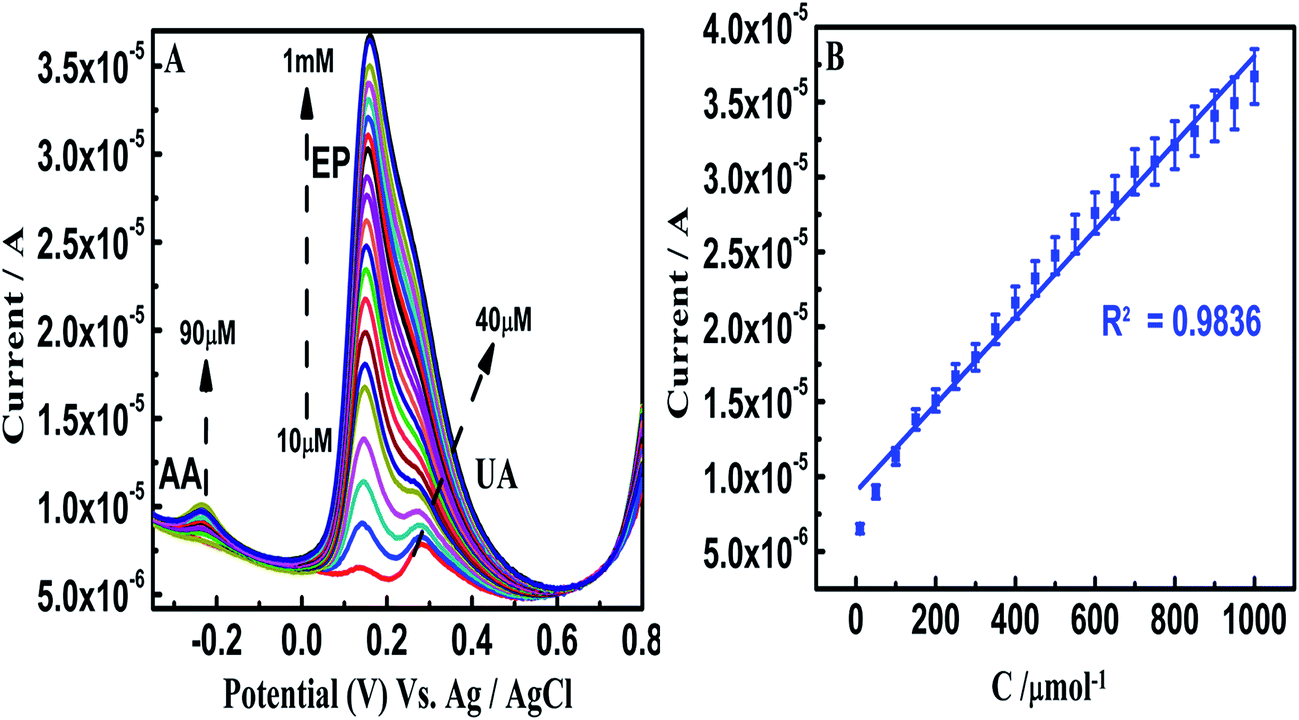 | ||
| Fig. 9 (A) SWV of different concentrations of EP 10 μM to 1 mM in the presence of fixed concentrations of AA and UA on β-NiS@rGO/AuNS/GCE in 0.1 M PBS (pH 7.0) (B) calibration plot for different concentrations of EP in the presence of AA and UA. | ||
 | ||
| Fig. 10 (A) SWV of different concentrations of UA 40 μM to 1 mM in the presence of fixed concentrations AA and EP on β-NiS@rGO/AuNS/GCE in 0.1 M PBS (pH 7.0) (B) calibration plot for different concentrations of UA in the presence of AA and UA. | ||
Simultaneous determination of these analytes is achieved on β-NiS@rGO/AuNS/GCE in the range of 1 μM to 1 mM, 2 μM to 1 mM, and 100 nM to 1 mM with detection limits of 682 nM, 1.3 μM and 6 nM, respectively (Fig. 11); whereas on β-NiS@rGO/GCE in the ranges 1–600 μM, 100–600 μM, and 1–600 μM for AA, EP and UA, respectively (Fig. S5(D)†). It is also clearly seen that the AuNS-modified GCE exhibits a comparatively wider detection range. Our fabricated sensor shows lower detection limits and wider linear ranges towards the oxidation of AA, EP and UA than those reported previously, owing to the excellent AuNS conductivity and electrocatalytic activity of β-NiS@rGO (ESI Table 1†). To the best of our knowledge, our hybrid material only shows the lowest detection limit of 6 nM for UA in the study of simultaneous determination of AA, EP and UA. Therefore, the β-NiS@rGO/AuNS hybrid is an advanced and remarkable electrode material, which may have promising applications in pharmaceutical and biomedical fields.
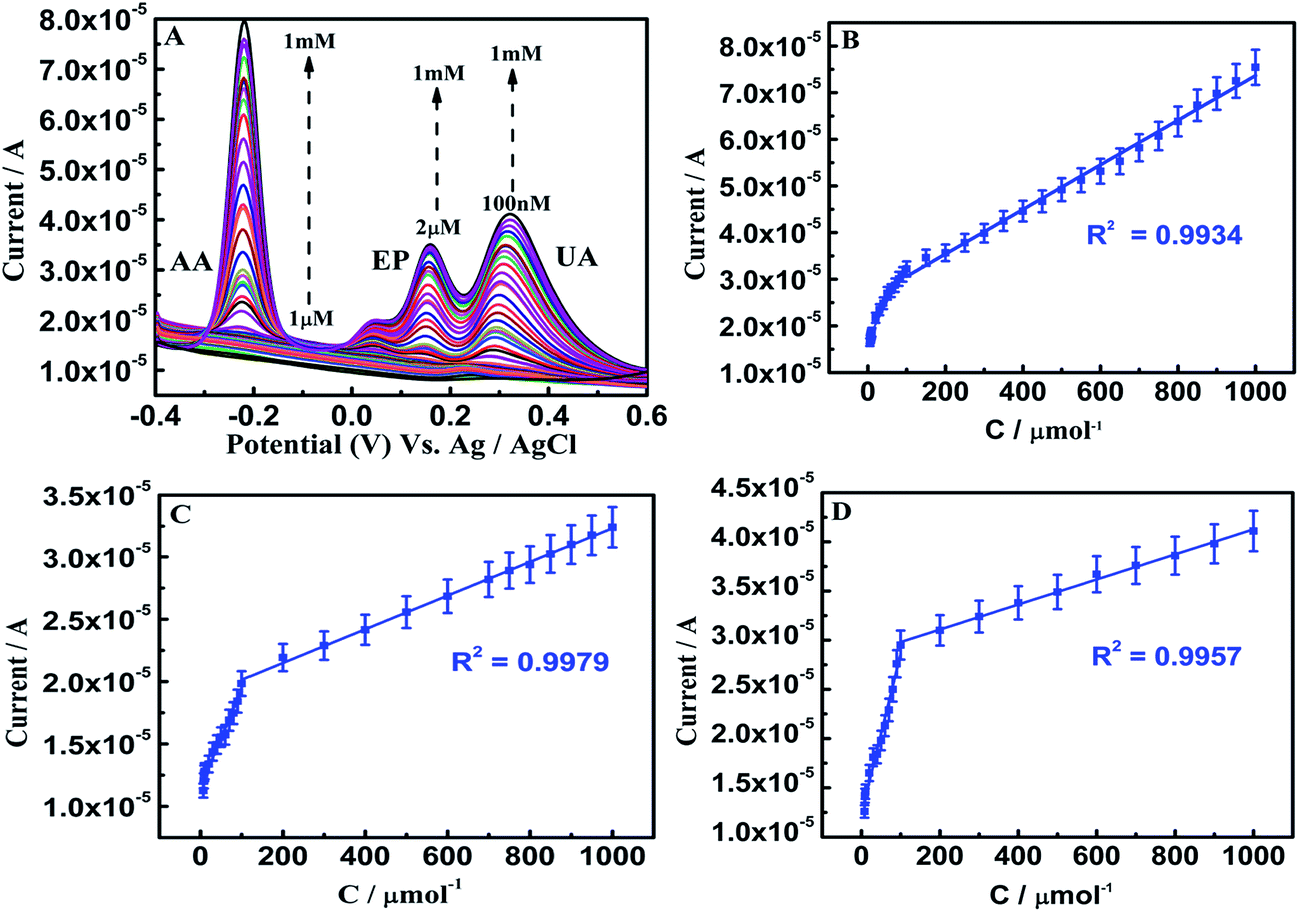 | ||
| Fig. 11 (A) SWV curves of AA, EP, and UA of different concentrations on β-NiS@rGO/AuNS/GCE in 0.1 M PBS (pH 7.0); AA 1 μM to 1 mM, EP 2 μM to 1 mM, and 100 nM to 1 mM UA. (B–D) Calibration plots of the oxidation peak currents against the concentrations of AA, EP, and UA. The error bars represent standard deviations for five tests. | ||
3.6. Interference study and stability of β-NiS@rGO/AuNS-modified GCE
We performed the selectivity test on our fabricated sensor using modified β-NiS@rGO/AuNS/GCE toward the oxidation of AA, EP and UA, each at 10 μM, with interfering foreign molecules added to the 0.1 M PBS solution (pH 7.0). The influence of these substances on the determination is evaluated, as shown in Fig. S6.† No interference is observed even in the presence of 1 mM of KCl, Na2SO4, NaNO3, glucose, citric acid and cysteine. These results show that β-NiS@rGO/AuNS hybrid possesses excellent selectivity for the determination of the three bio-compounds AA, EP and UA. The stability studies are also presented here, as shown in Fig. S4.† When one test of simultaneous determination toward the three analytes was completed, the sensor was kept in 0.1 M PBS (pH 7.0) and stored at 10 °C. After one month, the response peak currents of AA, EP and UA decreased by 6%, 5.5%, and 5.4%, respectively. In addition, the stability was also investigated by estimating the current responses of six independent β-NiS@rGO/AuNS/GCEs fabricated by the same procedure in 0.1 M PBS for 50 consecutive cycles at the scan rate 50 mV s−1 in the presence of 1 mM AA, EP and UA in 0.1 M PBS (pH 7.0). The electrode retained its initial response satisfactorily. To ascertain the reproducibility of the biosensor proposed, eight different GCEs were modified with the β-NiS@rGO/AuNS hybrid and their responses towards the oxidation of 100 μM each of AA, EP and UA were tested. The peak current obtained in the measurements of eight independent electrodes showed a relative standard deviation of 2.39%, 2.04% 1.49% for AA, EP and UA, respectively, confirming that the results are reproducible.4. Practical applications
Every real sample has a particular concentration of biomolecules present and we initially measured their SWV response. Then, a known different concentration of the analyte was added and the corresponding SWV response signals were measured. The observed current responses for the real samples were compared with those of the analytes whose concentration was known. The details of the real sample analyses are as follows:(i) For AA analysis in vitamin C with biotin tablet (100 mg per tablet content) the current response was in the range 8–450 μM; for lemon extract the current of AA was found in the range of 8–600 μM (Fig. S7(A) and (B)†).
(ii) 100 μL EP injection solution in 10 mL of 0.1 M PBS was taken and SWV exhibited the current values in the concentration range 1 μM to 1.25 mM (Fig. S7(C)†).
(iii) The urine samples were diluted 10 times with 0.1 M PBS solution (pH 7.0) without any other treatment before measurement. While adding the diluted urine sample 1, the peaks appeared in the range of 100 nM to 800 μM, and for sample 2, peaks showed the range 1 μM to 1 mM (Fig. S7(D) and (E)†).
(iv) Serum depicted current responses in the range of 300 μM to 1 mM, in which EP and UA presence was noticed simultaneously (Fig. S7(F)†).
(v) In human blood, we observed the presence of AA, EP and UA simultaneously. The concentration ranges of AA (70–550 μM), EP (40 μM to 1 mM) and UA (7–400 μM) (Fig. S7(G)†) were recorded. All the real sample analysis results are summarized in ESI Table 2.†
5. Conclusion
A β-NiS@rGO intertwined nanosheets hybrid catalyst was synthesized using a new exfoliation method and AuNS was electrodeposited for simultaneous performance of AA, EP and UA. The rGO on the NiS develops more active sites and acts as an excellent electron conductor to efficiently transfer from the analytes to the active site of the composite material. Similarly, the AuNS also enhanced the detection of analytes by stimulating effective electron transfer phenomena. This is believed to have created a better communication between the electrode surface and sensing analyte. The β-NiS@rGO/AuNS/GCE hybrid material is a promising substitute for precious metals in the fabrication of biosensors and energy storage devices.Acknowledgements
The author J. W. gratefully acknowledges Board of Research in Nuclear Sciences (34(1)/14/34/2015-BRNS with ATC) for financial assistance.References
- J. Yan, S. Liu, Z. Zhang, G. He, P. Zhou, H. Liang, L. Tian, X. Zhou and H. Jiang, Colloids Surf., B, 2013, 111, 392–397 CrossRef CAS PubMed.
- X. Zhang, Y. C. Zhang and L. X. Ma, Sens. Actuators, B, 2016, 227, 488–496 CrossRef CAS.
- J. Jiang and X. Du, Nanoscale, 2014, 6, 11303–11309 RSC.
- H. Sun, J. Chao, X. Zuo, S. Su, X. Liu, L. Yuwen, C. Fanab and L. Wang, RSC Adv., 2014, 4, 27625–27629 RSC.
- L. C. S. Figueiredo-Filho, T. A. Silva, F. C. Vicentini and O. Fatibello-Filho, Analyst, 2014, 139, 2842–2849 RSC.
- S. Yu, C. Luo, L. Wang, H. Peng and Z. Zhu, Analyst, 2013, 138, 1149–1155 RSC.
- S. Wang, W. Zhang, X. Zhong, Y. Chai and R. Yuan, Anal. Methods, 2015, 7, 1471–1477 RSC.
- S. Mehdi Ghoreishi, M. Behpour, S. Mousavi, A. Khoobi and F. S. Ghoreishi, RSC Adv., 2014, 4, 37979–37984 RSC.
- Y. Zhang, Y. J. Ziying Wang, S. Liu and T. Zhang, RSC Adv., 2015, 5, 106307–106314 RSC.
- T. H. Tsai, S. Thiagarajan, S. M. Chen and C. Y. Cheng, Thin Solid Films, 2012, 520, 3054–3059 CrossRef CAS.
- S. Priyatharshni, M. Divagar, C. Viswanathan, D. Mangalaraj and N. Ponpandian, J. Electrochem. Soc., 2016, 163, B460–B465 CrossRef CAS.
- N. Mahmood and Y. L. Hou, Adv. Sci., 2014, 1, 120 Search PubMed.
- S. B. Ni, X. L. Yang and T. Li, J. Mater. Chem., 2012, 22, 2395–2397 RSC.
- J. Jang, S. Jeong, J. W. Seo, M. C. Kim, E. Sim, Y. Oh, S. Nam, B. Park and J. Cheon, J. Am. Chem. Soc., 2011, 133, 7636–7639 CrossRef CAS PubMed.
- N. Mahmood, C. Z. Zhang and Y. L. Hou, Small, 2013, 9, 1321–1328 CrossRef CAS PubMed.
- C. X. Xu, Q. G. Zhai, Y. J. Liu, K. J. Huang, L. Lu and L. Ke-Xin, Anal. Bioanal. Chem., 2014, 406, 6943–6951 CrossRef CAS PubMed.
- C. Wei, C. Cheng, Y. Cheng, Y. Wang, Y. Xu, W. Du and H. Pang, Dalton Trans., 2016, 45, 10789–10797 RSC.
- Y. J. Yang, Sens. Actuators, B, 2015, 221, 750–759 CrossRef CAS.
- S. F. Kong, Z. T. Jin, H. Liu and Y. Wang, J. Phys. Chem. C, 2014, 118, 25355–25364 CAS.
- C. Z. Ma, J. Xu, J. Alvarado, B. H. Qu, J. Somerville, J. Y. Lee and Y. S. Men, Chem. Mater., 2015, 27, 5633–5640 CrossRef CAS.
- D. B. Kong, H. Y. He, Q. Song, B. Wang, W. Lv, Q. H. Yang and L. J. Zhi, Energy Environ. Sci., 2014, 7, 3320–3325 CAS.
- H. Geng, S. F. Kong and Y. Wang, J. Mater. Chem. A, 2014, 2, 15152–15158 CAS.
- G. Yan, Y. Xu and Y. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 801–806 Search PubMed.
- H. Bi, W. Zhao, S. Sun, H. Cui, T. Lin, F. Huang, X. Xie and M. Jiang, Carbon, 2013, 61, 116–123 CrossRef CAS.
- H. C. Tao, X. L. Yang, L. L. Zhang and S. B. Ni, J. Phys. Chem. Solids, 2014, 75, 1205–1209 CrossRef CAS.
- Z. Li, J. Han, L. Fan and R. Guo, CrystEngComm, 2015, 17, 1952–1958 RSC.
- F. Cao, R. Liu, L. Zhou, S. Y. Song, Y. Lei, W. Dong Shi, F. Zhaoab and H. Zhang, J. Mater. Chem., 2010, 20, 1078–1085 RSC.
- H. J. Kim, H. D. Lee, S. Srinivasa Rao, A. E. Reddy, S. K. Kim and C. V. Thulasi Varma, RSC Adv., 2016, 6, 29003–29019 RSC.
- L. Mi, Y. Chen, W. Wei, W. Chen, H. Hou and Z. Zheng, RSC Adv., 2013, 3, 17431–17439 RSC.
- A. Singh, A. J. Roberts, R. C. T. Sladeb and A. Chandra, J. Mater. Chem. A, 2014, 2, 16723–16730 CAS.
- C. Wei, C. Cheng, J. Zhao, Z. Wang, H. Wu, K. Gu, W. Du and H. Pang, ChemistryOpen, 2015, 4, 32–38 CrossRef CAS PubMed.
- G. Li, H. Huo and C. Xu, J. Mater. Chem. A, 2015, 23, 4922–4930 Search PubMed.
- S. Radhakrishnan and S. J. Kim, RSC Adv., 2015, 5, 44346–44352 RSC.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS PubMed.
- K. P. Loh, Q. L. Bao, P. K. Ang and J. X. Yang, J. Mater. Chem., 2010, 20, 2277–2289 RSC.
- H. L. Wang and H. J. Dai, Chem. Soc. Rev., 2013, 42, 3088–3113 RSC.
- S. Singh, S. K. Tuteja, D. Sillu, A. Deep and C. Raman Suri, Microchim. Acta, 2016, 183, 1729–1738 CrossRef CAS.
- K. C. Hung, Y. H. Lai and T. W. Lin, Catal. Sci. Technol., 2016, 6, 4020–4026 CAS.
- N. I. Kovtyukhova, P. J. Ollivier, B. R. Martin, T. E. Mallouk, S. A. Chizhik, E. V. Buzaneva and A. D. Gorchinskiy, Chem. Mater., 1999, 3, 771–778 CrossRef.
- D. Pan, L. Guo, J. Zhang, C. Xi, Q. Xue, H. Huang, J. Li, Z. Zhang, W. Yu, Z. Chen, Z. Li and M. Wu, J. Mater. Chem., 2012, 22, 3314 RSC.
- B. Li, H. Cao, J. Yin, Y. A. Wu and J. H. Warner, J. Mater. Chem., 2012, 22, 1876–1883 RSC.
- D. Pan, S. Wang, B. Zhao, M. Wu, H. Zhang, Y. Wang and Z. Jiao, Chem. Mater., 2009, 21, 3136–3142 CrossRef CAS.
- W. Li, S. Wang, L. Xin, M. Wu and X. Lou, J. Mater. Chem. A, 2016, 4, 7700–7709 CAS.
- H. Li, L. Chai, X. Wang, X. Wu, G. Xi, Y. Liu and Y. Qian, Cryst. Growth Des., 2007, 7, 1918–1922 CAS.
- F. Cai, R. Sun, Y. Kang, H. Chen, M. Chen and Q. Li, RSC Adv., 2015, 5, 23073–23079 RSC.
- K. Ding, H. Yang, Y. Wang and Z. Guo, Int. J. Electrochem. Sci., 2012, 7, 4663–4672 CAS.
- C. Sumathi, P. Muthukumaran, S. Radhakrishnan, G. Ravi and J. Wilson, RSC Adv., 2015, 5, 17888–17896 RSC.
- B. Goki Eda and M. Chhowalla, Adv. Mater., 2010, 22, 2392–2415 CrossRef PubMed.
- X. Shen, J. Sun, G. Wang, J. Park and K. Chen, Mater. Res. Bull., 2010, 45, 766–771 CrossRef CAS.
- Y. Zheng, A. Wang, H. Lin, L. Fu and W. Cai, RSC Adv., 2015, 5, 15425–15430 RSC.
- G. T. Soon How, A. Pandikumar, H. N. Ming and L. H. Ngee, Sci. Rep., 2014, 4, 5044 Search PubMed.
- Q. Sun, Vib. Spectrosc., 2009, 51, 213 CrossRef CAS.
- R. Akbarzadeh, H. Dehghani and F. Behnoudnia, Dalton Trans., 2014, 43, 16745–16753 RSC.
- J. Wang, B. Chen and B. Xing, Environ. Sci. Technol., 2016, 50, 3798–3808 CrossRef CAS PubMed.
- S. Zhang, D. Zhang, V. I. Sysoev, O. V. Sedelnikova, I. P. Asanov, M. V. Katkov, H. Song, V. Okotrub, L. G. Bulusheva and X. Chen, RSC Adv., 2014, 4, 46930–46933 RSC.
- Q. Pan, J. Xie, T. G. Zhu Cao, X. Zhao and S. Zhang, Inorg. Chem., 2014, 53, 3511–3518 CrossRef CAS PubMed.
- L. Mi, Q. Ding, W. Chen, Li. Zhao, H. Hou, C. Liu, C. Shen and Z. Zheng, Dalton Trans., 2013, 42, 5724–5730 RSC.
- X. Zuo, R. Zhang, B. Yang, G. Li, H. Tang, H. Zhang, M. Wu, Y. Ma, S. Jin and K. Zhu, J. Mater. Sci.: Mater. Electron., 2015, 26, 8176–8181 CrossRef CAS.
- C. Rajkumar, B. Thirumalraj, S. M. Chen and S. Palanisamy, RSC Adv., 2016, 6, 68798–68805 RSC.
- X. Niu, M. Lan, H. Zhao and C. Chen, Anal. Chem., 2013, 85, 3561–3569 CrossRef CAS PubMed.
- S. Mao, G. H. Lu, K. H. Yu, Z. Bo and J. H. Chen, Adv. Mater., 2010, 22, 3521–3526 CrossRef CAS PubMed.
- H. F. Zarick, O. Hurd, J. A. Webb, C. Hungerford, W. R. Erwin and R. Bardhan, ACS Photonics, 2014, 1, 806–811 CrossRef CAS.
- Y. Lou, S. Yuan, Y. Zhao, P. Hu, Z. Wang, M. Zhang, L. Shi and D. Li, Dalton Trans., 2013, 42, 5330–5337 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra19921f |
| This journal is © The Royal Society of Chemistry 2016 |