
Highly recyclable and magnetic catalyst of a metalloporphyrin-based polymeric composite for cycloaddition of CO2 to epoxide
Aibing Chena,
Pengpeng Juab,
Yunzhao Zhangab,
Jinzhu Chen*bc,
Hui Gaob,
Limin Chend and
Yifeng Yua
aCollege of Chemical and Pharmaceutical Engineering, Hebei University of Science and Technology, Shijiazhuang 050018, PR China
bGuangzhou Institute of Energy Conversion, Chinese Academy of Sciences, Guangzhou 510640, PR China. E-mail: chenjz@ms.giec.ac.cn; Fax: +86-20-3722-3380; Tel: +86-20-3722-3380
cSchool of Chemistry and Materials Science, Jinan University, Guangzhou 510632, PR China
dGuangdong Provincial Key Laboratory of Atmospheric Environment and Pollution Control, School of Environment and Energy, South China University of Technology, Guangzhou Higher Education Mega Centre, Guangzhou 510006, PR China
First published on 4th October 2016
Abstract
A new core-double-shell microsphere [Fe3O4@SiO2@Zn(Por)OP] was designed and prepared by coating a core–shell composite of Fe3O4 magnetic core and SiO2 shell (Fe3O4@SiO2) with a zinc porphyrin-based organic polymer [Zn(Por)OP]. While, Zn(Por)OP was readily obtained by a convenient condensation of pyrrole and terephthaldehyde in propanoic acid followed by a direct metallation with zinc acetate. Due to the presence of the inner core–shell particles (Fe3O4@SiO2), the cage construction of this composite microsphere Fe3O4@SiO2@Zn(Por)OP was significantly different from that of solid polymeric microsphere Zn(Por)OP, which further infected its physical properties to achieve a controllable morphology regulation easily. Besides, removing the inner core–shell particles of Fe3O4@SiO2 from Fe3O4@SiO2@Zn(Por)OP led to the formation of the Zn(Por)OP hollow microspheres which exhibited outstanding catalytic performance for the cycloaddition of carbon dioxide and propylene oxide to give propylene carbonate (PC) with turnover frequency (TOF) of 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 978 molPC molZn−1 h−1. For the Fe3O4@SiO2@Zn(Por)OP composite, the outer organic shell Zn(Por)OP served as the catalytically active layer for the cycloaddition and a PC yield of 97% was obtained. More importantly, the catalyst could be easily recycled from the reaction mixture by magnetic separation and no significant loss of the activity was observed after sixteen cycles. Therefore, this composite microsphere can provide insightful information for further design of magnetic materials with controllable morphology regulation as well as can be extended toward the development of highly efficient, stable and recyclable catalysts in other catalytic reaction systems.
978 molPC molZn−1 h−1. For the Fe3O4@SiO2@Zn(Por)OP composite, the outer organic shell Zn(Por)OP served as the catalytically active layer for the cycloaddition and a PC yield of 97% was obtained. More importantly, the catalyst could be easily recycled from the reaction mixture by magnetic separation and no significant loss of the activity was observed after sixteen cycles. Therefore, this composite microsphere can provide insightful information for further design of magnetic materials with controllable morphology regulation as well as can be extended toward the development of highly efficient, stable and recyclable catalysts in other catalytic reaction systems.
Introduction
As attractive catalytic materials, metalloporphyrins and their derived complexes have widely been investigated for cycloaddition of carbon dioxide (CO2) and epoxide to give cyclic carbonates, due to their outstanding catalytic properties and electron-deficient nature.1–9 The cycloaddition reactions of CO2 to epoxides are currently an important strategy for both chemical fixation of CO2 and production of carbonates. In addition to metalloporphyrins, metallosalens and ionic liquids have also shown excellent catalytic performance for the cycloaddition reaction. To heterogenize these catalytically active components for catalyst recycling, salen-Co/Al/Zn/Cr complex-derived microporous organic polymers,10–12 ionic liquid-grafted organic polymers,13 and polystyrene-supported quaternized ammonium salts14 were reported as excellent heterogeneous catalysts for the cycloaddition reactions. Besides, inspired by the excellent work on the chemical fixation of CO2 with metalloporphyrin-based metal–organic frameworks [M(Por)MOF],15–24 we reported a more promising candidate using metalloporphyrin-based organic polymers [M(Por)OP (M = Zn, Co)] which were, for the first time, used as heterogeneous catalysts for the synthesis of cyclic carbonates from epoxides and CO2.25 These polymeric catalysts, derived from metalloporphyrin, metallosalen and ionic liquid; while showing the advantages of easy tailoring and high accessibility, are very attractive for the coupling CO2 with epoxides. However, the design of these soft polymer-based catalysts with controllable morphology regulation should be more attractive not only to improve their catalytic performance for the cycloaddition reactions but also to reduce the loading level of the catalysts. While, the construction of magnetically polymeric composites should be more promising for workup method by easily and rapidly magnetic separation.On the other hand, cyclic carbonates, the cycloaddition productions, are widely employed as solvent, electrolyte and plastics softener.26–32 In the last several decades, in order to synthesize cyclic carbonate from CO2 and epoxides, various catalysts have been developed, such as alkali metal salts, transition metal complexes, metal oxides, organic bases, quaternary ammonium salts, phosphonium salts, Lewis acids, ionic liquids, cellulose, Cs loaded zeolites and alumina, organometallic compounds, metalloporphyrin and so on.33–51 In addition, due to the synergistic effect of the binary catalytic system, a combination of metalloporphyrin with a suitable nucleophile such as 4-dimethylaminopyridine (DMAP) or phenyltrimethylammonium tribromide (PTAT) can lead to the synthesis of the cyclic carbonate much more accessible, even under mild reaction conditions.52–55 In the research of heterogenization of metalloporphyrin complexes, heterogeneous catalysts such as biogenous iron oxide-, magnetic ferriferous oxide- and NaX zeolite-supported metalloporphyrin complexes were developed for the coupling reactions of CO2 with epoxides to yield carbonates.56–58 The advances were significant, however, most of these catalyst systems generally suffered from low catalytic activity or requirement high pressure and/or high temperature. Therefore, design of highly effective catalyst for chemical fixation of CO2 to form cyclic carbonates is still desirable.
Considering the ease and rapidity for the separation, magnetic core–shell Fe3O4@SiO2 particles with super magnetism property and stability can be a promising choice as support.57,59–63 Very recently, Fe-porphyrin networks on the magnetic supports Fe3O4 were used for catalytic carbene insertion into N–H bonds,64 which further inspired us to explore metalloporphyrin-based organic polymer with the Fe3O4@SiO2 as the inner core. Herein, we prepared the core–shell SiO2@(Por)OP and core-double-shell Fe3O4@SiO2@(Por)OP by convenient condensation of pyrrole and terephthaldehyde in the presence of SiO2 and Fe3O4@SiO2 particles in propanoic acid, respectively (Scheme 1).25 The (Por)OP hollow microspheres were also investigated by removing the inner SiO2 from the SiO2@(Por)OP with NH4·HF2 (Scheme 1). Moreover, SiO2@Zn(Por)OP, Fe3O4@SiO2@Zn(Por)OP and Zn(Por)OP hollow microspheres were readily achieved by direct metallation SiO2@(Por)OP, Fe3O4@SiO2@(Por)OP and (Por)OP hollow microspheres with zinc acetate, respectively. The presence of the inner SiO2 and Fe3O4@SiO2 make for the cage construction of the composites SiO2@(Por)OP and Fe3O4@SiO2@(Por)OP different from that of solid polymeric microspheres (Por)OP, which further infects their physical properties. Our newly prepared catalysts SiO2@Zn(Por)OP, Fe3O4@SiO2@Zn(Por)OP and Zn(Por)OP hollow microspheres were performed in accordance with our own efforts toward the development of highly efficient, stable, recyclable and heterogeneous catalyst for cycloaddition of CO2 to epoxides with the outer organic shell Zn(Por)OP serving as the catalytically active layer (Scheme 1 and Fig. 1). Moreover, the catalysts were insoluble in any commonly used reaction media and could be easily separated from the reaction system and reused. We believe that the efficient and recyclable catalytic system has great potential application. Under the optimal reaction conditions, a propylene carbonate (PC) yield of 97% was obtained by using Fe3O4@SiO2@Zn(Por)OP catalyst with KI as co-catalyst for the cycloaddition reaction of CO2 to propylene oxide (PO, Fig. 1). More importantly, Fe3O4@SiO2@Zn(Por)OP can be readily reused at least for sixteen times without significant loss of activity. Besides, the Zn(Por)OP hollow microspheres exhibited efficient catalytic performance for the cycloaddition reaction with the turnover frequency (TOF) of 13978 molPC molZn−1 h−1.
 | ||
| Scheme 1 Schematic illustration for the preparation of Zn(Por)OP-based catalysts and the proposed structures of Zn(Por)OP, SiO2@Zn(Por)OP, Zn(Por)OP hollow microspheres and Fe3O4@SiO2@Zn(Por)OP. | ||
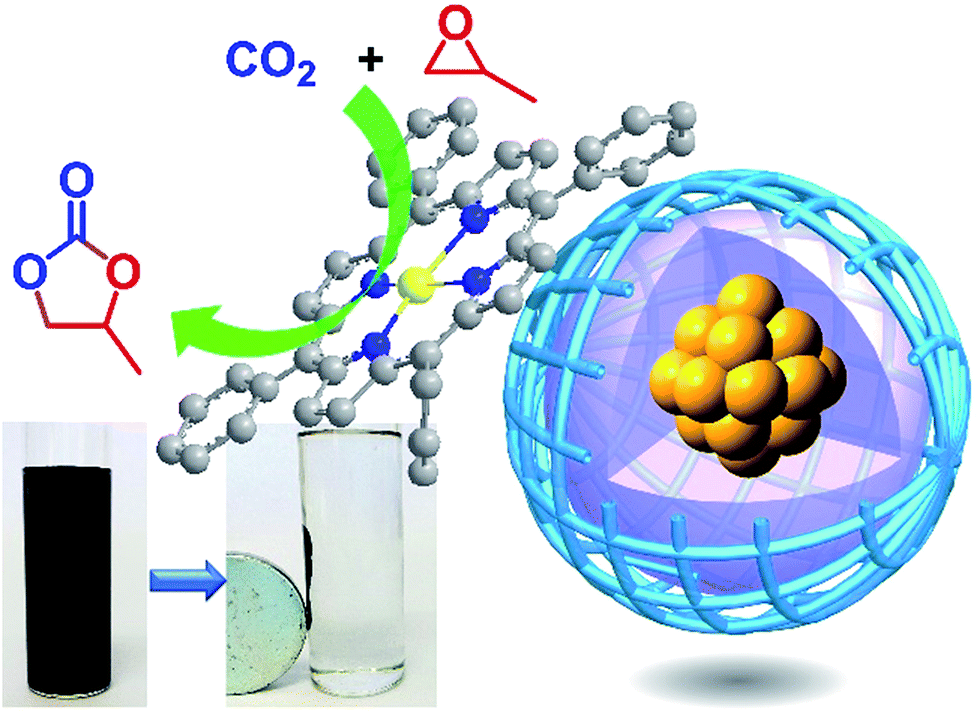 | ||
| Fig. 1 Magnetically separable Fe3O4@SiO2@Zn(Por)OP catalyst for cycloaddition of CO2 to PO. | ||
Experimental section
Materials
Unless otherwise stated, all chemicals in this research were commercial available and used without further purification. Propylene oxide (PO), zinc acetate [Zn(OAc)2·2H2O], propanoic acid (98 wt%), ethanol, methanol, dichloromethane, tetrahydrofuran (THF), N,N-dimethylformamide (DMF), NH4·HF2 were obtained from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, P. R. China). Terephthaldehyde, pyrrole, potassium iodide (KI), potassium bromide (KBr), tetraphenylphosphonium bromide (TPPB), tetrabutylammonium bromide (TBAB), Fe3O4 (mean nanoparticles size 20 nm), SiO2 (mean nanoparticles size 500 nm) were purchased from Aladdin Industrial Inc. (Shanghai, P. R. China). Pyrrole was freshly distilled before use. Carbon dioxide (CO2 > 99.999%) was obtained from Huate Co. Ltd. (Foshan, P. R. China).Synthesis of silica-coated magnetite nanoparticles (Fe3O4@SiO2)
Fe3O4 (2.0 g) with average diameter of 20 nm suspended in a mixture of ethanol (35 mL) and distilled water (6 mL) was sonicated for 15 min. Tetraethyl orthosilicate (TEOS, 1.5 mL) was then added slowly to the mixture and sonicated for another 10 min. Then aqueous ammonia (10%, 1.4 mL) was added slowly over 10 min under mechanical stirring and the mixture was then heated at 40 °C for 12 h. The iron oxide nanoparticles with a thin layer of silica (Fe3O4@SiO2) were separated by an external magnetic field, washed three times with ethanol and then dried under vacuum.65Synthesis of Fe3O4@SiO2@(Por)OP core-double-shell microspheres
Fe3O4@SiO2@(Por)OP was prepared by a direct condensation of pyrrole and terephthaldehyde in the presence of Fe3O4@SiO2 in propanoic acid. Fe3O4@SiO2 (200 mg) were dispersed in propanoic acid (370 mL) with sonication for 20 min. Subsequently, terephthaldehyde (300 mg) and freshly distilled pyrrole (150 mg) mixed with 30 mL propanoic acid were added dropwisely into the above solution in 10 minutes under vigorous stirring and refluxing conditions. The mixture was further stirred for 3 h and then cooled down to room temperature. The Fe3O4@SiO2@(Por)OP was separated by an external magnetic field, washed thoroughly with distilled water, methanol, THF, dichloromethane and then vacuum dried in an oven at 80 °C overnight. Finally, the Fe3O4@SiO2@(Por)OP material was rigorously washed by Soxhlet extractions for 24 h with water, dichloromethane, methanol, and tetrahydrofuran, respectively, and then vacuum dried in an oven at 80 °C for another 12 h.Synthesis of SiO2@(Por)OP core–shell microspheres
SiO2@(Por)OP was prepared following the same synthetic procedure used for Fe3O4@SiO2@(Por)OP except that Fe3O4@SiO2 (200 mg) was replaced by SiO2 (100 mg) with average diameter of 500 nm.Synthesis of (Por)OP hollow microspheres
(Por)OP hollow microspheres was prepared by soaking the core–shell structure SiO2@(Por)OP composites in an aqueous solution of NH4·HF2 (25 wt%) with vigorous stirring for 48 h and then the product was washed with distilled water and ethanol. Finally, the (Por)OP hollow spheres were dried in an oven at 80 °C for 12 h.Synthesis of SiO2@Zn(Por)OP
SiO2@Zn(Por)OP was prepared by a direct metallation of SiO2@(Por)OP with Zn(OAc)2. SiO2@(Por)OP (500 mg) and Zn(OAc)2·2H2O (1.50 g) were added to a mixture of dried DMF/propanoic acid (400 mL, v/v = 2:1). After refluxing for 5 h at 140 °C, the solution was cooled down to room temperature and filtered. The filter cake was washed thoroughly with methanol and hot water alternately. Finally, the black filter cake was dried under vacuum at 80 °C overnight to remove absorbed propanoic acid and other solvents.
Synthesis of Fe3O4@SiO2@Zn(Por)OP and Zn(Por)OP hollow microspheres
Fe3O4@SiO2@Zn(Por)OP and Zn(Por)OP hollow microspheres were prepared following the same synthetic procedure as for SiO2@Zn(Por)OP.General procedure for coupling reaction of CO2 and PO
In a typical experimental procedure, the cycloaddition of CO2 to PO was carried out in a 25 mL stainless steel autoclave equipped with a magnetic stir bar. The autoclave reactor was successively charged with catalyst Fe3O4@SiO2@Zn(Por)OP (61 mg, 0.023 mmol Zn, 0.00046 mol% Zn relative to PO), co-catalyst KI (1.0 mmol, 2.0 mol% relative to PO) and PO (2.904 g, 50.0 mmol). After purging the reactor several times with CO2, the outlet valve was then closed to maintain the pressure at 3.0 MPa (room temperature) in the system controlled by a large CO2 gas reservoir which was connected to the reaction cell. The reaction was performed at a stirring speed of 600 rpm at 120 °C for 2.0 h. The reactor was cooled down to room temperature with ice-water bath. The remained mixture was filtered and distilled under reduced pressure to obtain pure PC.Characterization techniques
Fourier transform infrared (FT-IR) spectra of samples with KBr wafers were recorded at room temperature in the 500–4500 cm−1 region with a Bruker Tensor 27 spectrometer equipped with a Data Station at a spectral resolution of 1 cm−1 and accumulations of 128 scans. The surface morphology and structures of SiO2@Zn(Por)OP, Fe3O4@SiO2@Zn(Por)OP, Zn(Por)OP hollow microspheres were investigated by scanning electron microscopy (SEM) on a FESEM Hitachi-S 4800. The samples were mounted on an aluminum stub using adhesive carbon tape and SEM image were obtained at various magnifications. The morphological analysis was carried out using transmission electron microscopy (TEM, JEM-2100HR). Samples for TEM studies were prepared by placing a drop of the suspension of SiO2@Zn(Por)OP and Zn(Por)OP hollow microspheres samples in ethanol onto a carbon-coated copper grid, followed by evaporating the solvent. X-ray photoelectron spectroscopy (XPS) spectra was performed with a Kratos Axis Ultra (DLD) photoelectron spectrometer operated at 15 kV and 10 mA at a pressure of about 5 × 10−9 torr using AlKα as the exciting source (hν = 1486.6 eV). C 1s photoelectron peak (BE = 284.2 eV) was used for the binding energy calibration. Metal contents in samples were determined quantitatively by inductively coupled plasma-atomic emission spectroscopy (ICP-AES) analysis on an IRIS Advantage 1000 instrument. Thermogravimetry (TGA) and differential thermal analyses (DTA) of the SiO2@Zn(Por)OP, Fe3O4@SiO2@Zn(Por)OP, Zn(Por)OP hollow microspheres were carried out in a TGA Instruments thermal analyzer TA-SDT Q-600. 13C{1H} NMR spectra of (Por)OP hollow microspheres and Zn(Por)OP hollow microspheres were recorded on a Bruker Avance 300 solid-state NMR spectrometer at 75 MHz for 13C.Results and discussion
Metalloporphyrin-based organic polymers
Scheme 1 shows the synthetic route for Zn(Por)OP-derived various catalysts. Polymer (Por)OP was prepared by a condensation of terephthaldehyde and pyrrole in propanoic acid; while, metalloporphyrin-based organic polymer Zn(Por)OP was obtained by subsequent metallation (Por)OP with Zn(OAc)2.25 Zn(Por)OP was composed of solid and spherical microparticles. In this research, (Por)OP- and Zn(Por)OP-coated SiO2 [SiO2@(Por)OP and SiO2@Zn(Por)OP] were obtained following the same synthetic procedure as for (Por)OP and Zn(Por)OP, respectively, but in the presence of silica microparticles (SiO2) (Scheme 1). Fig. 2a shows the scanning electron microscopy (SEM) images of silica microparticles used as the core in the synthesis of core–shell composite. The silica particles showed well mono-dispersity and were all in spherical shape with the mean diameter about 500 nm. In the case of SiO2@Zn(Por)OP, as shown in the SEM images (Fig. 2b and c), the shape of the composite microparticles was still in spherical and the particles showed a narrow size distribution. It means that Zn(Por)OP shell enwrapped the silica microparticle core effectively and formed the spherical silica core–metalloporphyrin shell composite microparticles. Transmission electron microscopy (TEM) images provided further insights into the structure of SiO2@Zn(Por)OP. Herein, the SiO2 cores, homogeneously surrounded by zinc porphyrin-based organic shell with average coating thickness around 120 nm, can be clearly observed (Fig. 2g–i). To further investigate the Zn(Por)OP shell on the surface of silica microspheres, the SiO2@(Por)OP was treated with NH4·HF2 to completely etch the inner SiO2 affording (Por)-OP hollow microspheres. Subsequent metallation of (Por)OP hollow microspheres with Zn(OAc)2 led to the formation of Zn(Por)OP hollow microspheres (Scheme 1). Herein, SiO2 functions as hard template for the formation of Zn(Por)OP hollow microspheres. Fig. 2j–l show typical TEM image of Zn(Por)OP hollow microspheres with shell thickness. The sharp contrast between the dark edge and the pale center in the TEM image further proved the hollow inside. The shell thicknesses of the hollow Zn(Por)OP were around 120–150 nm, which was in line with the thickness of the Zn(Por)OP shell over the surface of SiO2@Zn(Por)OP. Notably, the hollow polymeric Zn(Por)OP maintained rigid structure without any evident distortions from a spherical shape despite the presence of the hollow inner space in the polymer and the soft nature of polymeric layer (Fig. 2f and j–l). Finally, Zn(Por)OP-derived magnetic catalyst [Fe3O4@SiO2@Zn(Por)OP] was synthesized by coating silica-coated magnetite nanoparticles (Fe3O4@SiO2) with a layer of Zn(Por)OP. Fig. 2d shows the SEM images of Fe3O4@SiO2@Zn(Por)OP, it can be seen that the materials were all in spherical shape and the surfaces of the composite microparticles were rough. The SEM images also indicated the successful coating of the magnetic Fe3O4 particles. As shown in Fig. 2e, there was none breakdown of the spherical morphology of the particles even after sixteen times recycling for the cycloaddition reaction (Fig. 11), which proved the catalysts were firm in structure. Moreover, due to the magnetic nature of Fe3O4@SiO2@Zn(Por)-OP, the catalyst can readily be retrieved from the reaction system by using a magnet as shown in Fig. 1. | ||
| Fig. 2 The SEM images of (a) SiO2 core microparticles, (b and c) SiO2@Zn(Por)OP, (d and e) Fe3O4@SiO2@Zn(Por)OP, and (f) Zn(Por)OP hollow microspheres; TEM images of (g–i) for SiO2@Zn(Por)OP, and (j–l) for Zn(Por)OP hollow microspheres. | ||
The solid state 13C CP-MAS NMR spectra of (Por)OP hollow microspheres and Zn(Por)OP hollow microspheres were shown in Fig. 3. Zn(Por)OP hollow microspheres exhibited four resonances from 135 to 110 ppm (Fig. 3). The signals at 135 and 130 ppm were assigned to the phenylene linkages; while, the peaks at 120 and 110 ppm were indexed to porphyrin macrocycles.25,66,67 The solid state 13C CP-MAS NMR spectra of (Por)OP hollow microspheres were very close to those of Zn(Por)OP hollow microspheres (Fig. 3).
 | ||
| Fig. 3 Solid-state 13C CP/MAS NMR spectra of (Por)OP hollow microspheres and Zn(Por)OP hollow microspheres recorded at a MAS rate of 12 kHz. | ||
Fourier transform infrared (FT-IR) spectra of SiO2, Fe3O4@SiO2@Zn(Por)OP, SiO2@Zn(Por)OP and Zn(Por)OP hollow microspheres were shown in Fig. 4. In the absence of SiO2, the bands appear in the Zn(Por)OP hollow spheres at the 3022–2835 cm−1 can be assigned to aromatic C–H stretching modes from pyrrole and benzene rings. However, except for the relevant peaks about SiO2, the spectra of Fe3O4@SiO2@Zn(Por)OP, SiO2@Zn(Por)OP and Zn(Por)OP hollow microspheres were closely resemble each other. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CC stretching vibrations of the porphyrin macrocycle and benzene ring led to five absorption peaks in the region from 1600 to 1400 cm−1.68,69 The band at 1005 cm−1 was metal-dependent and could be ascribed to an in-plane porphyrin deformation mode due to the coordination of Zn(II) with porphyrin units. The similar phenomenon was also previously observed for the divalent metal derivatives of the tetraphenylporphines.22,70 The absorption band near 803 cm−1 can be attributed to out-of-plane deformations of the substituted phenyl ring. The FT-IR analyses clearly verified that a porphyrin macrocycle segment existed in the materials of Fe3O4@SiO2@Zn(Por)OP, SiO2@Zn(Por)OP and Zn(Por)OP hollow microspheres.
N and CC stretching vibrations of the porphyrin macrocycle and benzene ring led to five absorption peaks in the region from 1600 to 1400 cm−1.68,69 The band at 1005 cm−1 was metal-dependent and could be ascribed to an in-plane porphyrin deformation mode due to the coordination of Zn(II) with porphyrin units. The similar phenomenon was also previously observed for the divalent metal derivatives of the tetraphenylporphines.22,70 The absorption band near 803 cm−1 can be attributed to out-of-plane deformations of the substituted phenyl ring. The FT-IR analyses clearly verified that a porphyrin macrocycle segment existed in the materials of Fe3O4@SiO2@Zn(Por)OP, SiO2@Zn(Por)OP and Zn(Por)OP hollow microspheres.
 | ||
| Fig. 4 FT-IR spectra of SiO2, Fe3O4@SiO2@Zn(Por)OP, SiO2@Zn(Por)-OP and Zn(Por)OP hollow microspheres. | ||
To examine the surface compositions, X-ray photoelectron spectroscopy (XPS) spectra of Fe3O4@SiO2@(Por)OP and Fe3O4@SiO2@Zn(Por)OP were performed. Fig. 5a shows full range XPS survey spectra including carbon and nitrogen for both Fe3O4@SiO2@(Por)OP and Fe3O4@SiO2@Zn(Por)OP, zinc for Fe3O4@SiO2@Zn(Por)OP. The N(1s) spectrum peak of Fe3O4@SiO2@(Por)OP was deconvoluted into two superimposed peaks at 397.9 and 399.8 eV, corresponding to iminic N (–NC–) and pyrrolic N (–N–H–), respectively,68–75 which suggests the presence of porphyrin macrocycle in the prepared Fe3O4@SiO2@(Por)OP (Fig. 5b). Compared with Fe3O4@SiO2@(Por)OP, an additional N(1s) peak of Fe3O4@SiO2@Zn(Por)OP was observed, which can be attributed to the transfer of electron density from N (–NC–) to the Zn ion, suggesting the formation of N–Zn bond.76 Moreover, the Zn(2p) XPS spectrum of Fe3O4@SiO2@Zn(Por)OP showed two main peaks located at 1044.9 and 1021.8 eV corresponding to Zn(2p1/2) and Zn(2p3/2), respectively, suggesting the presence of Zn(II)-centered porphyrin units in Fe3O4@SiO2@Zn(Por)OP (Fig. 5c).73 To probe catalyst stability, the recovered Fe3O4@SiO2@Zn(Por)OP after sixteen-time recycling was further characterized with XPS. The N(1s) XPS of recovered Fe3O4@SiO2@Zn(Por)OP was deconvoluted into three peaks at 397.9, 399.4 and 399.9 eV and was very close to the combined spectra of Fe3O4@SiO2@(Por)OP and Fe3O4@SiO2@Zn(Por)OP (Fig. 5b), indicating the presence of iminic N (–NC–), coordinated N (–NC–) to the Zn ion, and pyrrolic N (–N–H–) structures. This observation can be attributed to a partial leaching of Zn(II) species leading to the formation of porphyrin units. Both fresh and recovered Fe3O4@SiO2@Zn(Por)OP show almost identical two peaks in the Zn(2p) XPS spectrum (Fig. 5c); however, the zinc content decreased from 0.24% for the fresh Fe3O4@SiO2@Zn(Por)OP to 0.19% for the recovered one after sixteen recycles based on XPS analysis. To probe the thermal stabilities of these Zn(Por)OP-derived materials, Fe3O4@SiO2@Zn(Por)OP, SiO2@Zn(Por)OP and Zn(Por)OP hollow microspheres were performed with thermal gravimetric-differential thermal analysis (TG-DTA). The curves of weight loss versus temperature for Zn(Por)OP-derived materials show no significant weight loss before 200 °C.
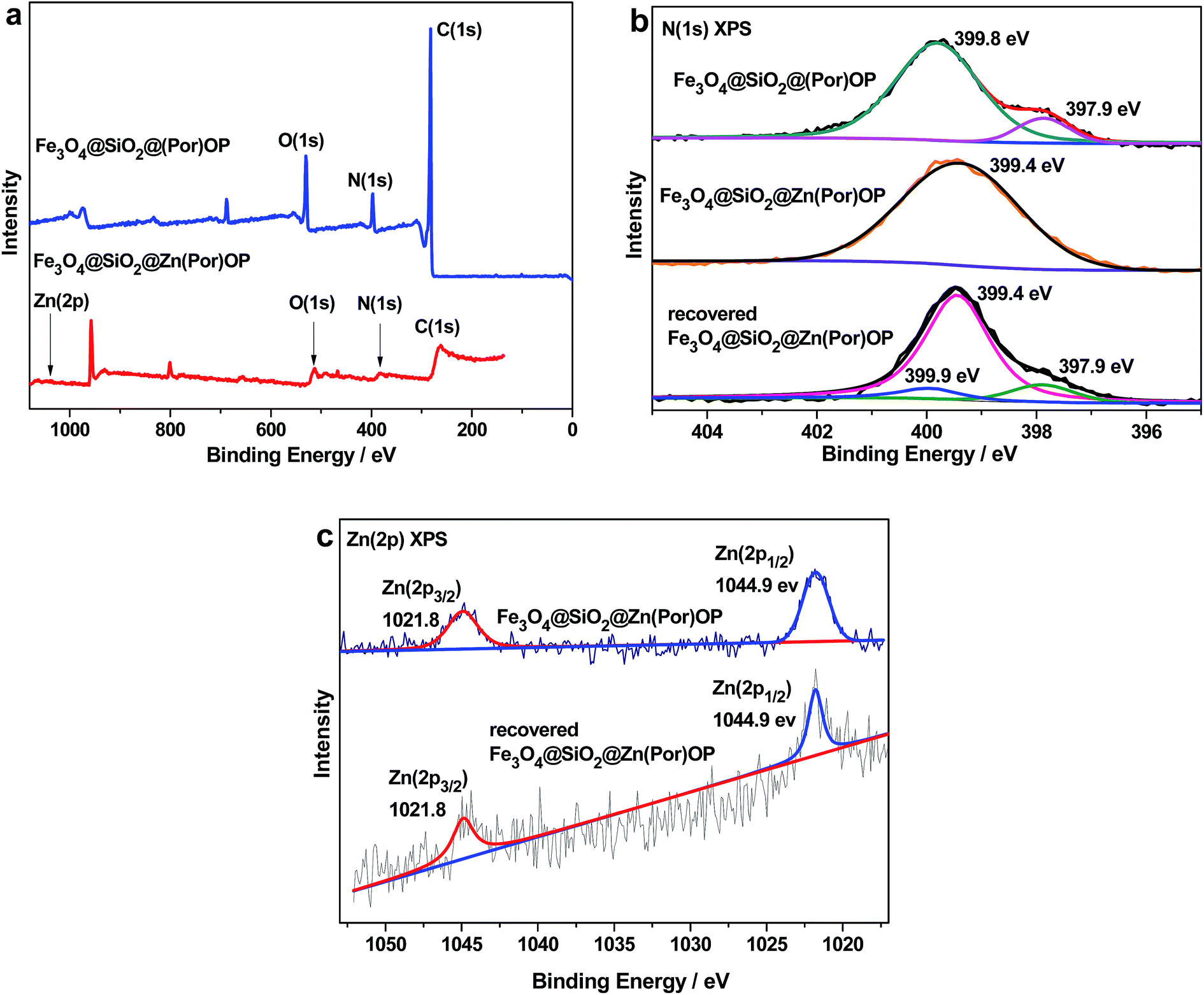 | ||
| Fig. 5 (a) XPS scan survey of Fe3O4@SiO2@(Por)OP and Fe3O4@SiO2@Zn(Por)OP; (b) N(1s) XPS of Fe3O4@SiO2@(Por)OP, fresh and recovered Fe3O4@SiO2@Zn(Por)OP; (c) Zn(2p) XPS of fresh and recovered Fe3O4@SiO2@Zn(Por)OP. | ||
Cycloaddition of CO2 to PO
Zn(Por)OP-derived heterogeneous catalysts including SiO2@Zn(Por)OP, Zn(Por)OP hollow microspheres, and Fe3O4@SiO2@Zn(Por)OP were thus investigated for the coupling reaction of CO2 and PO to give PC. While, co-catalysts such as potassium bromide (KBr), potassium iodide (KI), tetraphenylphosphonium bromide (TPPB), and tetrabutylammonium bromide (TBAB) were carefully chosen as nucleophilic reagents. The cycloaddition reaction of CO2 to PO was promoted by a combination of various catalysts and co-catalysts under solvent free conditions in a stainless autoclave.Fig. 6a compared the catalytic performance of Zn(Por)OP-based bi-component system for the cycloaddition reaction. The catalytic activity of the catalysts SiO2@Zn(Por)OP, Zn(Por)-OP hollow microspheres, and Fe3O4@SiO2@Zn(Por)OP were very low in the absence of co-catalyst with PC yields ranging from 4% to 6% which were, however, slightly high than blank SiO2@(Por)OP producing PC yield of 2%. In the case of co-catalysts, both KBr and KI were unreactive towards the cycloaddition yielding PC yields of 0.6% and 1.5%, respectively, under the investigated conditions. As phosphonium salt, co-catalyst TPPB, however, showed low catalytic activity when used alone in the cycloaddition reaction, presumably due to its low solubility in the PO. However, with the cycloaddition reaction proceeding, the product PC can play a polar aprotic solvents and TPPB will be free soluble in it. So drawing on the previous experience, it was proved that TPPB alone could directly catalyze the chemical fixation of CO2 onto epoxides. Therefore, TPPB showed remarkably enhanced activity in the bi-component catalytic system. Notably, co-catalyst TBAB was catalytically active towards the cycloaddition giving PC yield 70% in the present experiments.
 | ||
| Fig. 6 Screening of the catalytic performance of Zn(Por)OP-based catalytic systems for the synthesis of cyclic PC from CO2 and PO in terms of (a) PC yield and (b) turnover frequency (TOF). Reaction conditions: (a) PC (2.904 g, 50.0 mmol), catalyst [SiO2@Zn(Por)OP (40 mg), Zn(Por)OP hollow microspheres (37 mg), Fe3O4@SiO2@Zn(Por)OP (61 mg), SiO2@(Por)OP (40 mg), 0.023 mmol Zn, 0.00046 mol% Zn relative to PO], co-catalyst [KI, KBr, TPPB, TBAB (1.0 mmol, 2.0 mol% relative to PO)], CO2 (3.0 MPa), 120 °C, 2 h, in a 25 mL autoclave; (b) catalyst [SiO2@Zn(Por)OP, Fe3O4@SiO2@Zn(Por)OP, Zn(Por)OP hollow microspheres], co-catalyst [KI, TPPB, KBr, TBAB (44 equivalents to catalyst)], CO2 (3.0 MPa), 120 °C. | ||
Obviously, co-catalyst such as KI and TPPB can significantly promote the catalytic activity of the catalysts including SiO2@Zn(Por)OP, Zn(Por)OP hollow microspheres, and Fe3O4@SiO2@Zn(Por)OP in the cycloaddition. Moreover, the combinations of SiO2@Zn(Por)OP, Zn(Por)OP hollow microspheres, and Fe3O4@SiO2@Zn(Por)OP with KI showed a similar catalytic activity in terms of PC yields (95–97%). This phenomenon showed that Lewis acid site of Zn2+ and nucleophilic reagent I− played a key role on the coupling reaction of CO2 and PO. Moreover, the combination of catalyst with KI was considerably more active to afford PC than the combination of the corresponding catalyst with KBr. For example, the highest yield of PC was obtained by 97% when SiO2@Zn(Por)OP and KI were used to catalyze the reaction. However, negligible PC yield of 2.6% was observed from SiO2@Zn(Por)OP/KBr, due to cooperation of the relatively strong nucleophilicity and the moderate leaving ability of I− relative to those of Br−.77,78
The catalytic performances of SiO2@Zn(Por)OP, Zn(Por)OP hollow microspheres, Fe3O4@SiO2@Zn(Por)OP were further compared in terms of TOF for the cycloaddition reactions (Fig. 6b). Herein, the TOF of PO-to-PC transformation was measured at the PO conversion ranging from 10% to 20%, given as moles of PC produced per mole of Zn per hour for the catalyst. For example, when the coupling reactions were performed at 120 °C using Zn(Por)OP/KI-based bi-component system as the catalysts, Fig. 6b shows a decreased TOF for PO-to-PC transformation with the order of Zn(Por)OP hollow microspheres (2699 molPC molZn−1 h−1) > Fe3O4@SiO2@Zn(Por)OP (2001 molPC molZn−1 h−1) > SiO2@Zn(Por)OP (1949 molPC molZn−1 h−1) > Zn(Por)OP (1628 molPC molZn−1 h−1). Besides, the co-catalyst type also played a key role on the PC formation, and the promotion effect of various co-catalyst was further quantitatively compared in terms of TOF as depicted in Fig. 6b. For instance, when the coupling reactions were performed at 120 °C using Zn(Por)OP hollow microspheres/co-catalyst-based bi-component system, a decreased TOF for PO-to-PC transformation with the order of TPPB (13978 molPC molZn−1 h−1) > TBAB (12772 molPC molZn−1 h−1) > KI (2699 molPC molZn−1 h−1) > KBr (231 molPC molZn−1 h−1). Notably, the bi-component system of Zn(Por)OP hollow microspheres/TPPB was considerable active to afford PC with the measured TOF of 13978 molPC molZn−1 h−1 under the investigated conditions, which was significantly high than in the cases of any other combinations.
The influence of temperature on the yield of PC was investigated with SiO2@Zn(Por)OP catalyst as shown in Fig. 7a. An increase in temperature resulted in an increase in the PC yield. Especially, the PC yield increased sharply when temperature increasing from 70 to 100 °C. Moreover, the reaction temperature also significantly affected the catalytic performance in terms of TOF for PO-to-PC transformation. As shown in Fig. 7b, the TOFs increased from 368 molPC molZn−1 h−1 at 70 °C to 8158 molPC molZn−1 h−1 at 190 °C with SiO2@Zn(Por)OP/KI as the catalysts, confirming that raising the reaction temperature dramatically promoted the coupling reaction. Besides, when reaction temperature was below 140 °C, the catalyst SiO2@Zn(Por)-OP did not show any visible differences from Zn(Por)OP. However, with the temperature increased above 140 °C, the difference was significant. For example, the TOFs increased from 3699 molPC molZn−1 h−1 at 140 °C to 8158 molPC molZn−1 h−1 at 190 °C using SiO2@Zn(Por)OP/KI as catalyst. While, the TOFs just increased from 2637 molPC molZn−1 h−1 at 140 °C to 3183 molPC molZn−1 h−1 at 190 °C using Zn(Por)OP/KI as catalyst.25
 | ||
| Fig. 7 Effect of reaction temperature on the cycloaddition reaction of CO2 to PO in terms of (a) PC yield and (b) TOF using SiO2@Zn(Por)OP catalyst. Reaction conditions: (a) PO (2.904 g, 50.0 mmol), SiO2@Zn(Por)OP (40 mg, 0.023 mmol Zn, 0.00046 mol% Zn relative to PO), KI (166 mg, 1.0 mmol), CO2 (3.0 MPa), 70–160 °C, 2 h, in a 25 mL autoclave; (b) catalyst [SiO2@Zn(Por)OP], KI [44 equivalents to SiO2@Zn(Por)OP], 70–190 °C, CO2 (3.0 MPa). | ||
The influence of the initial CO2 pressure on the yield of PC was also checked with SiO2@Zn(Por)OP catalyst. With the increase of CO2 pressure from 0.5 to 4.0 MPa, the PC yield significantly increased from 48% to 97% (Fig. 8a). The pressure effect on the catalytic performance of SiO2@Zn(Por)OP in terms of TOFs indicated that the influence was more significant (Fig. 8b). The TOFs decreased significantly from 2816 to 2050 molPC molZn−1 h−1 with an increase in pressure from 1.0 to 2.0 MPa. The TOFs remained nearly constant at initial CO2 pressure in a small range of 2.0 to 3.0 MPa. With the further increase the pressure, the TOFs decreased to 1429 molPC molZn−1 h−1 with the CO2 pressure of 4.0 MPa. This phenomenon could be due to the acidic nature of CO2 and basic property of PO, increasing CO2 pressure promoted the formation of CO2–PO complex rather than improved the interactions between PO and the catalyst, thus leading to a decreased catalytic performance in terms of TOFs.13,79–82
 | ||
| Fig. 8 Effect of initial CO2 pressure on the cycloaddition reaction of CO2 to PO in terms of (a) PC yield and (b) TOF using SiO2@Zn(Por)OP catalyst. Reaction conditions: (a) PO (2.904 g, 50.0 mmol), SiO2@Zn(Por)OP (40 mg, 0.023 mmol Zn, 0.00046 mol% Zn relative to PO), KI (166 mg, 1.0 mmol), CO2 (0.5–4.0 MPa), 120 °C, 2 h, in a 25 mL autoclave; (b) catalyst [SiO2@Zn(Por)OP], KI [44 equivalents to SiO2@Zn(Por)OP], 120 °C, 2 h, CO2 pressure (0.5–4.0 MPa). | ||
Fig. 9 demonstrates the dependence of the PC yield on reaction time at 120 °C under CO2 pressure of 3.0 MPa using SiO2@Zn(Por)OP catalyst. The results indicated that the yield increased to 98% with reaction time within 2.0 h. Then the yield increased slightly with prolonged reaction time. Therefore, under the optimized reaction conditions, SiO2@Zn(Por)OP/KI-promoted cycloaddition of CO2 to PO provided PC yield 97% at 120 °C after 2 h reaction time under 3.0 MPa CO2 atmospheric pressure (Fig. 8 and 9).
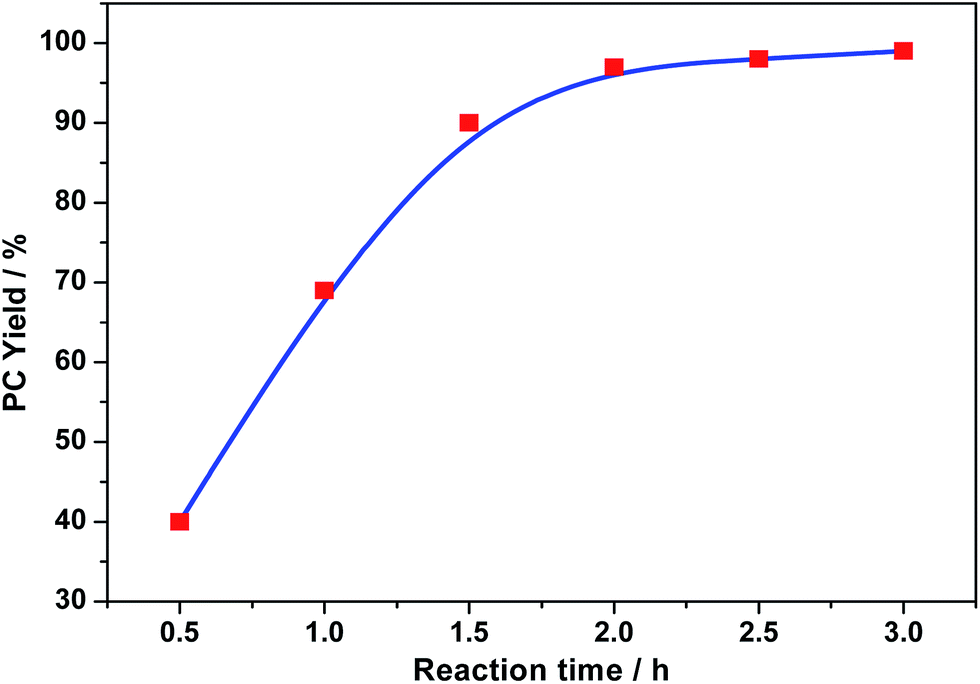 | ||
| Fig. 9 Influence of reaction time on the cycloaddition reaction of CO2 to PO using SiO2@Zn(Por)OP catalyst. Reaction conditions: PO (2.904 g, 50.0 mmol), catalyst [SiO2@Zn(Por)OP (40 mg), 0.023 mmol Zn, 0.00046 mol% Zn relative to PO], KI (166 mg, 1.0 mmol), CO2 (3.0 MPa), 120 °C, 0.5–3.0 h, in a 25 mL autoclave. | ||
In the case of magnetic catalyst, Fig. 10 shows the influence of reaction temperature, CO2 pressure and reaction time on the cycloaddition reaction of CO2 to PO with Fe3O4@SiO2@Zn(Por)OP catalyst. Generally, the effects of reaction conditions on PO-to-PC transformation for Fe3O4@SiO2@Zn(Por)OP exhibited similar tendency when compared with SiO2@Zn(Por)OP. Under the optimized reaction conditions, Fe3O4@SiO2@Zn(Por)OP/KI-promoted cycloaddition of CO2 to PO provided PC yield 98% at 120 °C after 2 h reaction time under 3.0 MPa CO2 atmospheric pressure (Fig. 10).
 | ||
| Fig. 10 Influence of (a) reaction temperature, (b) CO2 pressure and (c) reaction time on the cycloaddition reaction of CO2 to PO using magnetic Fe3O4@SiO2@Zn(Por)OP catalyst. Reaction conditions: PO (2.904 g, 50.0 mmol), catalyst [Fe3O4@SiO2@Zn(Por)OP (61 mg), 0.023 mmol Zn, 0.00046 mol% Zn relative to PO], KI (166 mg, 1.0 mmol) in a 25 mL autoclave, [CO2 (3.0 MPa), 2.0 h] for (a), [120 °C, 2.0 h] for (b) and [CO2 (3.0 MPa), 120 °C] for (c). | ||
Catalyst recycling
To probe the reusability of the magnetic catalyst Fe3O4@SiO2@Zn(Por)OP for the cycloaddition reactions, a sixteen-cycle experiment was performed under the optimized reaction conditions. The catalyst could be easily recovered via magnetic separation (Fig. 1), was reused for next run after rinsed with water, alcohol, dichloromethane, respectively, and then was dried on the vacuum. As shown in Fig. 11, high yields of PC were attained in all successive recycles without significant loss of activity after sixteen cycles, which indicated that the catalyst Fe3O4@SiO2@Zn(Por)OP exhibited excellent reusability and stability for the repetitive use in the cycloaddition reactions. The formation of highly stable catalyst was attributed to covalently and highly-cross linked catalytic sites of metalloporphyrin in the polymer matrices of the shell of Fe3O4@SiO2@Zn(Por)OP (Scheme 1 and Fig. 1). While, the formation of highly active catalyst was related to the dense catalytic sites of metalloporphyrin in the shell of Fe3O4@SiO2@Zn(Por)OP (Scheme 1 and Fig. 1). The slightly decreased catalytic performance of Fe3O4@SiO2@Zn(Por)OP in the recycling can presumably be related to the leaching of approximately 21% of the Zn from Fe3O4@SiO2@Zn(Por)OP after the sixteen-cycle experiment based on XPS analysis. | ||
| Fig. 11 Recycling of Fe3O4@SiO2@Zn(Por)OP catalyst. Reaction conditions: PO (2.904 g, 50.0 mmol), catalyst [Fe3O4@SiO2@Zn(Por)-OP (61 mg), 0.023 mmol Zn, 0.00046 mol% Zn relative to PO], co-catalyst [KI (1.0 mmol), 2.0 mol% relative to PO], CO2 (3.0 MPa), 120 °C, 2 h, in a 25 mL autoclave. | ||
Conclusions
In summary, a new microsphere [Fe3O4@SiO2@Zn(Por)OP] was designed and prepared, by coating core–shell magnetic microparticles of Fe3O4@SiO2 with the zinc metalloporphyrin-based organic polymer Zn(Por)OP, which showed efficient catalytic activities for cycloaddition of CO2 and propylene oxide. Due to the presence of the coated inner microparticles (Fe3O4@SiO2), the cage construction of this microsphere Fe3O4@SiO2@(Por)-OP is significantly different from that of (Por)OP, which further infects its physical properties to achieve the controllable morphology regulation easily. Under the optimal conditions, a propylene carbonate yield of 97% was obtained by using Fe3O4@SiO2@Zn(Por)OP catalyst with KI as co-catalyst. Most importantly, the catalytic systems could be easily recycled from the reaction mixture by magnetic separation and there were without significant loss of activity after sixteen cycles. Therefore, this microsphere catalytic system can provide insightful information for further design of magnetic materials with controllable morphology regulation as well as is helpful for the development of highly efficient, stable and recyclable catalysts in other catalytic reaction system.Acknowledgements
We are grateful for the financial support from National Natural Science Foundation of China (21172219, 21207039 and 21303209), Natural Science Foundation of Guangdong Province, China (2015A030312007) and Guangdong Provincial Key Laboratory of Atmospheric Environment and Pollution Control.References
- R. Srivastava, T. H. Bennur and D. Srinivas, J. Mol. Catal. A: Chem., 2005, 226, 199–205 CrossRef CAS.
- T. Aida and S. Inoue, J. Am. Chem. Soc., 1983, 105, 1304–1309 CrossRef CAS.
- W. Wu, X. F. Sheng, Y. S. Qin, L. J. Qiao, Y. Y. Miao, X. H. Wang and F. S. Wang, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2346–2355 CrossRef CAS.
- D. Feng, W. Chung, Z. Wei, Z. Gu, H. Jiang, Y. Chen, D. J. Darensbourg and H. Zhou, J. Am. Chem. Soc., 2013, 135, 17105–17110 CrossRef CAS PubMed.
- T. Aida and S. Inoue, Macromolecules, 1982, 15, 682–684 CrossRef CAS.
- W. J. Kruper and D. D. Dellar, J. Org. Chem., 1995, 60, 725–727 CrossRef CAS.
- F. Ahmadi, S. Tangestaninejad, M. Moghadam, V. Mirkhani, I. Mohammadpoor-Baltork and A. R. Khosropour, Inorg. Chem. Commun., 2011, 14, 1489–1493 CrossRef CAS.
- R. V. Siriwardane, M. Shen, E. P. Fisher and J. A. Poston, Energy Fuels, 2001, 15, 279–284 CrossRef CAS.
- L. Broussard and D. P. Shoemaker, J. Am. Chem. Soc., 1960, 82, 1041–1051 CrossRef CAS.
- Y. Xie, T.-T. Wang, X.-H. Liu, K. Zou and W.-Q. Deng, Nat. Commun., 2013, 4, 1960 Search PubMed.
- Y. Xie, T.-T. Wang, R.-X. Yang, N.-Y. Huang, K. Zou and W.-Q. Deng, ChemSusChem, 2014, 7, 2110–2114 CrossRef CAS PubMed.
- Y. Xie, R.-X. Yang, N.-Y. Huang, H.-J. Luo and W.-Q. Deng, J. Energy Chem., 2014, 23, 22–28 CrossRef CAS.
- Y. Xie, Z. F. Zhang, T. Jiang, J. L. He, B. X. Han, T. B. Wu and K. L. Ding, Angew. Chem., Int. Ed., 2007, 46, 7255–7258 CrossRef CAS PubMed.
- S.-D. Lee, B.-M. Kim, D.-W. Kim, M.-I. Kim, K. R. Roshan, M.-K. Kim, Y.-S. Won and D.-W. Park, Appl. Catal., A, 2014, 486, 69–76 CrossRef CAS.
- W.-Y. Gao, L. Wojtas and S. Q. Ma, Chem. Commun., 2014, 50, 5316–5318 RSC.
- M. H. Beyzavi, R. C. Klet, S. Tussupbayev, J. Borycz, N. A. Vermeulen, C. J. Cramer, J. F. Stoddart, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2014, 136, 15861–15864 CrossRef CAS PubMed.
- B. Saha, D. Gupta, M. M. Abu-Omar, A. Modak and A. Bhaumik, J. Catal., 2013, 299, 316–320 CrossRef CAS.
- A. M. Shultz, O. K. Farha, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2009, 131, 4204–4205 CrossRef CAS PubMed.
- R. K. Totten, Y.-S. Kim, M. H. Weston, O. K. Farha, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2013, 135, 11720–11723 CrossRef CAS PubMed.
- Y. Ferrand, P. L. Maux and G. Simonneaux, Tetrahedron: Asymmetry, 2005, 16, 3829–3836 CrossRef CAS.
- L. Chen, Y. Yang, Z. Q. Guo and D. L. Jiang, Adv. Mater., 2011, 23, 3149–3154 CrossRef CAS PubMed.
- L. Chen, Y. Yang and D. L. Jiang, J. Am. Chem. Soc., 2010, 132, 9138–9143 CrossRef CAS PubMed.
- A. Modak, J. Mondal and A. Bhaumik, Appl. Catal., A, 2013, 459, 41–51 CrossRef CAS.
- H. J. Mackintosh, P. M. Budd and N. B. McKeown, J. Mater. Chem., 2008, 18, 573–578 RSC.
- A. B. Chen, Y. Z. Zhang, J. Z. Chen, L. M. Chen and Y. F. Yu, J. Mater. Chem. A, 2015, 3, 9807–9816 CAS.
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, Angew. Chem., Int. Ed., 2002, 41, 2599–2602 CrossRef CAS.
- B. Schäffner, J. Holz, S. P. Verevkin and A. Börner, ChemSusChem, 2008, 1, 249–253 CrossRef PubMed.
- P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS PubMed.
- H. Sugimoto and K. Kuroda, Macromolecules, 2008, 41, 312–317 CrossRef CAS.
- J. Bayardon, J. Holz, B. Schäffner, V. Andrushko, S. Verevkin, A. Preetz and A. Börner, Angew. Chem., Int. Ed., 2007, 46, 5971–5974 CrossRef CAS PubMed.
- S. Mang, A. I. Cooper, M. E. Colclough, N. Chauhan and A. B. Holmes, Macromolecules, 2000, 33, 303–308 CrossRef CAS.
- L. M. Stamp, S. A. Mang, A. B. Holmes, K. A. Knights, Y. R. de Miguel and L. F. McConvey, Chem. Commun., 2001, 2502–2503 RSC.
- M. Mikkelsen, M. Jorgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 CAS.
- J. Sun, X. Q. Yao, W. G. Cheng and S. J. Zhang, Green Chem., 2014, 16, 3297–3304 RSC.
- T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312–1330 RSC.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC.
- J. Sun, W. G. Cheng, Z. F. Yang, J. Q. Wang, T. T. Xu, J. Y. Xin and S. J. Zhang, Green Chem., 2014, 16, 3071–3078 RSC.
- S. G. Liang, H. Z. Liu, T. Jiang, J. L. Song, G. Y. Yang and B. X. Han, Chem. Commun., 2011, 47, 2131–2133 RSC.
- M. Q. Zhu and M. A. Carreon, J. Appl. Polym. Sci., 2014, 131, 39738 Search PubMed.
- C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482–1497 CAS.
- C. J. Whiteoak and A. W. Kleij, Synlett, 2013, 24, 1748–1756 CrossRef CAS.
- N. Kielland, C. J. Whiteoak and A. W. Kleij, Adv. Synth. Catal., 2013, 355, 2115–2138 CrossRef CAS.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC.
- S. N. Riduan and Y. Zhang, Dalton Trans., 2010, 39, 3347–3357 RSC.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- D. J. Darensbourg and S. J. Wilson, Green Chem., 2012, 14, 2665–2671 RSC.
- Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956–9964 RSC.
- S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460–1479 CrossRef CAS.
- I. Omae, Coord. Chem. Rev., 2012, 256, 1384–1405 CrossRef CAS.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed.
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed.
- D. S. Bai, S. H. Duan, L. Hai and H. W. Jing, ChemCatChem, 2012, 4, 1752–1758 CrossRef CAS.
- T. Ema, Y. Miyazaki, S. Koyama, Y. Yano and T. Sakai, Chem. Commun., 2012, 48, 4489–4491 RSC.
- R. L. Paddock, Y. Hiyama, J. M. McKay and S. T. Nguyen, Tetrahedron Lett., 2004, 45, 2023–2026 CrossRef CAS.
- L. Jin, H. Jing, T. Chang, X. Bu, L. Wang and Z. Liu, J. Mol. Catal. A: Chem., 2007, 261, 262–266 CrossRef CAS.
- T. Ema, Y. Miyazaki, T. Taniguchi and J. Takada, Green Chem., 2013, 15, 2485–2492 RSC.
- D. Bai, Q. Wang, Y. Song, B. Li and H. Jing, Catal. Commun., 2011, 12, 684–688 CrossRef CAS.
- F. Y. Zhang, Y. J. Xie, P. L. Liu, F. Hao, Z. J. Yao and H. A. Luo, Catal. Lett., 2014, 144, 1894–1899 CrossRef CAS.
- A. Hu, G. T. Yee and W. Lin, J. Am. Chem. Soc., 2005, 127, 12486–12487 CrossRef CAS PubMed.
- F. Y. Yang, Y. Chu, S. Y. Ma, Y. P. Zhang and J. L. Liu, J. Colloid Interface Sci., 2006, 301, 470–478 CrossRef CAS PubMed.
- S. R. Jagtap, V. P. Raje, S. D. Samant and B. M. Bhanage, J. Mol. Catal. A: Chem., 2007, 266, 69–74 CrossRef CAS.
- Q. An, Z. F. Li, R. B. Graff, J. Guo, H. Gao and C. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 4969–4978 CAS.
- B. Fu, H. C. Yu, J. W. Huang, P. Zhao, J. Liu and L. N. Ji, J. Mol. Catal. A: Chem., 2009, 298, 74–80 CrossRef CAS.
- J. Yoo, N. Park, J. H. Park, J. H. Park, S. Kang, S. M. Lee, H. J. Kim, H. Jo, J.-G. Park and S. U. Son, ACS Catal., 2015, 5, 350–355 CrossRef CAS.
- B. Karimi, H. M. Mirzaei and E. Farhangi, ChemCatChem, 2014, 3, 758–762 CrossRef.
- L. Chen, Y. Yang and D. L. Jiang, J. Am. Chem. Soc., 2010, 132, 9138–9143 CrossRef CAS PubMed.
- A. Modak, M. Nandi, J. Mondal and A. Bhaumik, Chem. Commun., 2012, 48, 248–250 RSC.
- L. J. Boucher and J. J. Katz, J. Am. Chem. Soc., 1967, 89, 1340–1345 CrossRef CAS PubMed.
- G. D. Dorough, J. R. Miller and F. M. Huennekens, J. Am. Chem. Soc., 1951, 73, 4315–4320 CrossRef CAS.
- D. W. Thomas and A. E. Martell, J. Am. Chem. Soc., 1959, 81, 5111–5119 CrossRef CAS.
- J. M. Gottfried, K. Flechtner, A. Kretschmann, T. Lukasczyk and H.-P. Steinrück, J. Am. Chem. Soc., 2006, 128, 5644–5645 CrossRef CAS PubMed.
- D. K. Sarkar, X. J. Zhou, A. Tannous, M. Louie and K. T. Leung, Solid State Commun., 2003, 125, 365–368 CrossRef CAS.
- K. Flechtner, A. Kretschmann, L. R. Bradshaw, M.-M. Walz, H.-P. Steinrück and J. M. Gottfried, J. Phys. Chem. C, 2007, 111, 5821–5824 CAS.
- M. Röckert, S. Ditze, M. Stark, J. Xiao, H.-P. Steinrück, H. Marbach and O. Lytken, J. Phys. Chem. C, 2014, 118, 1661–1667 Search PubMed.
- W. Hieringer, K. Flechtner, A. Kretschmann, K. Seufert, W. Auwärter, J. V. Barth, A. Görling, H.-P. Steinrück and J. M. Gottfried, J. Am. Chem. Soc., 2011, 133, 6206–6222 CrossRef CAS PubMed.
- M. S. Killian, J.-F. Gnichwitz, A. Hirsch, P. Schmuki and J. Kunze, Langmuir, 2010, 26, 3531–3538 CrossRef CAS PubMed.
- A. M. B. do Rego, A. M. Ferraria and M. R. Vilar, J. Phys. Chem. C, 2013, 117, 22298–22306 Search PubMed.
- Q. Li, W. Zhang, N. Zhao, W. Wei and Y. Sun, Catal. Today, 2006, 115, 111–116 CrossRef CAS.
- J. H. Shi, J. L. Song, J. Ma, Z. F. Zhang, H. G. Fan and B. X. Han, Pure Appl. Chem., 2013, 8, 1633–1641 Search PubMed.
- T. Ema, Y. Miyazaki, S. Koyama, Y. Yano and T. Sakai, J. Am. Chem. Soc., 2014, 136, 15270–15279 CrossRef CAS PubMed.
- J. M. Sun, S. I. Fujita, F. Y. Zhao and M. Arai, Appl. Catal., A, 2005, 287, 221–226 CrossRef CAS.
- R. Nomura, M. Kimura, S. Teshima, A. Ninagawa and H. Matsuda, Bull. Chem. Soc. Jpn., 1982, 55, 3200–3207 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |