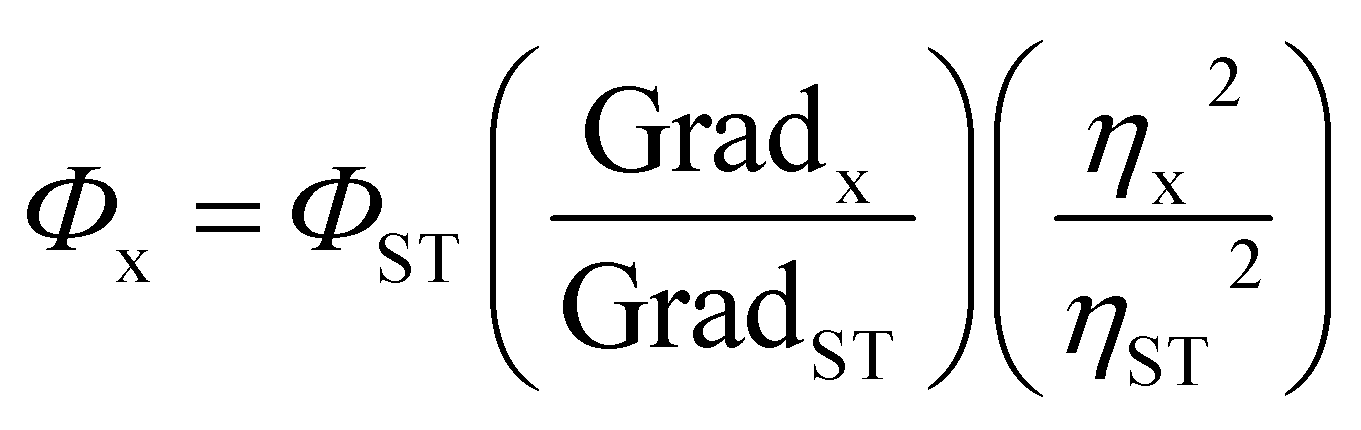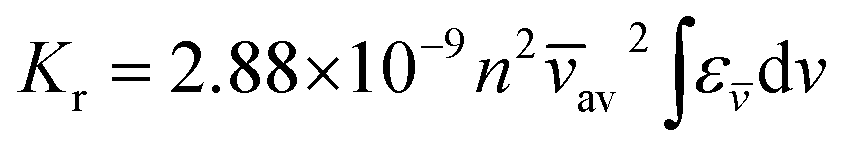DOI:
10.1039/C6RA19899F
(Paper)
RSC Adv., 2016,
6, 100271-100280
Novel triphenylamine based rhodamine derivatives: synthesis, characterization, photophysical properties and viscosity sensitivity†
Received
6th August 2016
, Accepted 17th October 2016
First published on 17th October 2016
Abstract
Five novel triphenylamine based deep red to NIR emitting rhodamine derivatives were synthesized and characterized by 1H and 13C NMR spectroscopy and elemental analysis. The photophysical properties of all these derivatives were studied in their spirocyclic as well as open form. Hydroxyl substituted derivatives were found to show red shifted emissions as compared to the plain rhodamine derivatives, while triphenylamine substituted derivatives showed larger Stokes shift and emission in the NIR region. All these newly synthesized rhodamine derivatives show comparatively larger Stokes shift (44–135 nm) than the commercially available Rhodamine B and Rhodamine 101. In their open form they are found to exhibit different emission color from pink (619 nm) to dark blue (719 nm) in day as well as UV-light. We also studied the interconversion of dye RH-2 from its spirocyclic to open form with the addition of acid (TFA in toluene). They are studied for their viscosity sensitivity and found to show very high fluorescence enhancement in polar viscous media such as ethanol–glycerol in their open form.
Introduction
Rhodamines belong to an important class of xanthene dyes, which are non-fluorescent and colorless in their spirocyclic form due to the small π-conjugated system, whereas highly fluorescent in their open form due to the elongated π-conjugated systems.1 The first synthesis of rhodamine was reported in 1905 by Noelting and Dziewoński.2 The ring opening mechanism of rhodamine derivatives by the addition of metal ion instead of acid was first time proposed by Czarnik and co-workers.3 Later on different rhodamine derivatives were synthesized and utilized for the sensing of variety of metal ions for e.g. lead,4 mercury,5–8 copper,9,10 iron,11–14 etc. Furthermore due to their excellent photophysical properties such as high molar extinction coefficient, excellent fluorescence quantum yield and very good photo stability, rhodamine dyes are used as laser dyes,15,16 in living cell imaging studies,17,18 as fluorescence standards for quantum yield calculation,19 as molecular switches20 and as fluorescent markers in biological studies.21 Recently rhodamine based energy transfer cassettes have been developed where rhodamine unit often acts as energy acceptor both in through space energy transfer (FRET)22–31 as well as through bond energy transfer.32–38 Although rhodamine dyes show versatile photophysical properties, most of the rhodamine derivatives absorb or emit below 600 nm making them non suitable for biological applications. Few researchers developed novel synthetic strategies to get longer wavelength rhodamine derivatives by replacing the central oxygen atom by other elements such as N, C, S, Se and Si.39–43 However synthesis of such type of NIR emitting dyes is tedious and multistep reactions. Also the most commonly used rhodamine dyes show very small Stokes shift (20–40 nm) which is again limiting its application for biological imaging in living systems due to auto fluorescence. There are a number of reports where the carboxylic acid group of rhodamine dyes is functionalized to acquire its different derivatives for their sensing as well as energy transfer applications but very few examples where the main rhodamine core is modified.44 There are very few protocols available on the modification of substituents including donating amine side nitrogen as well as extended conjugation on phenyl ring to acquire novel rhodamine dyes with red shifted absorption as well as emission and larger Stokes shift.45–49 Due to wide variety of applications and hitherto synthesis of the novel rhodamine dyes in the present studies showing absorption and emission wavelengths above 600 nm with large Stokes shift are highly desirable and worth considering. As triphenylamine is known for its unique donating ability and intramolecular charge transfer character, we designed and synthesized novel triphenylamine based rhodamine dyes which show red shifted absorption as well as emission and high Stokes shift.
A group of fluorescent molecules that consists of an electron donor group in π-conjugation with an electron acceptor group, also called as fluorescent molecular rotors [FMRs] and which forms twisted intramolecular charge transfer (TICT) states upon photo excitation have attracted much attention in recent years.50–52 Recently different triphenylamine derivatives are also found to show very good FMR properties due to the freely rotating phenyl rings around C–N bond which gets hindered in viscous environment causing fluorescence enhancement.53 As per our knowledge the rhodamine derivatives are not yet studied for their FMR properties. We studied the viscosity sensitivity of these triphenylamine based rhodamine derivatives with the increasing percentage of glycerol in ethanol solvent and observed that all these dyes show very good viscosity sensitivity in polar environment in their open form, particularly the RH-2 dye with 24 fold fluorescence enhancement.
Result and discussion
Synthetic strategy
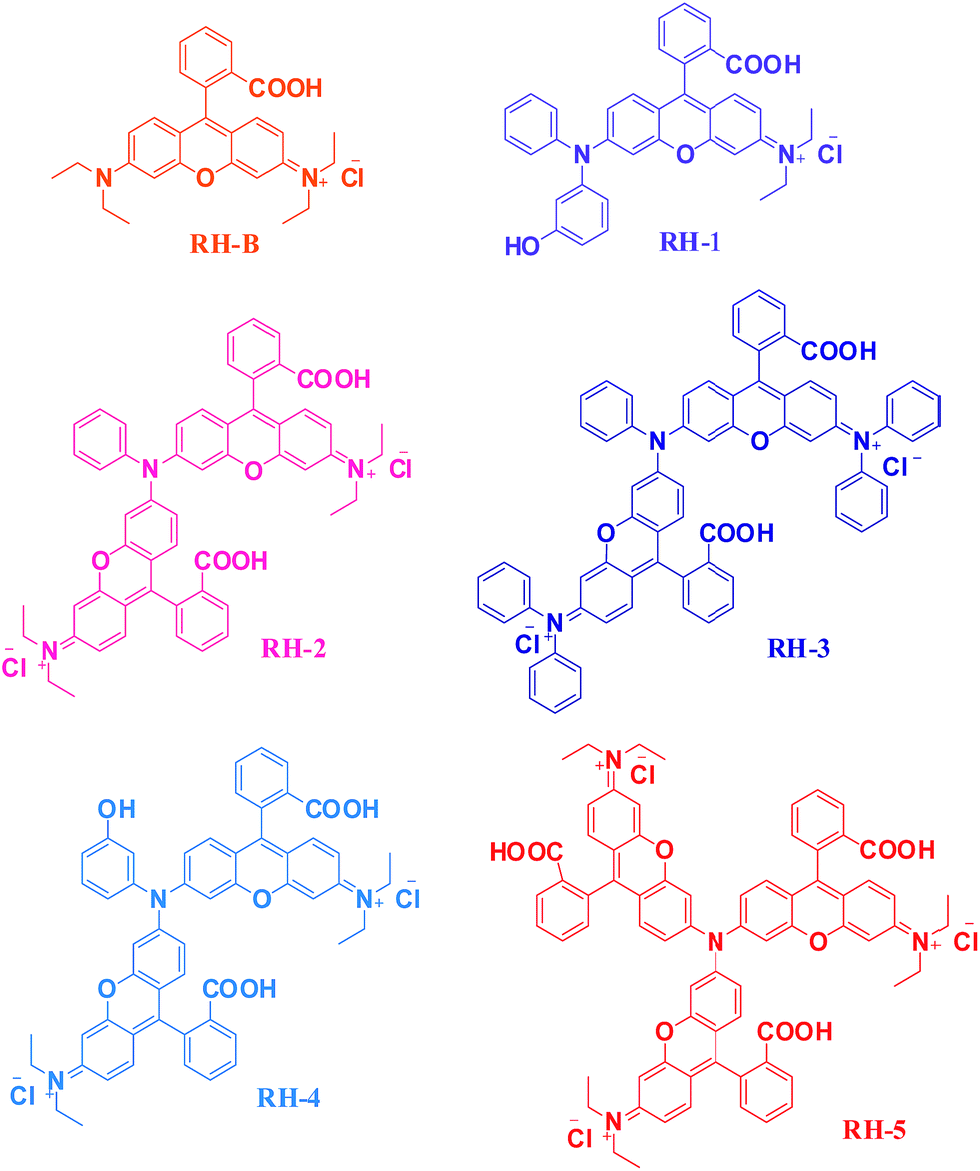
Intermediates 1, 2 and 5 were synthesized as per the procedure reported from our group.54 For the deprotection of methoxy intermediates 1 and 2 to their respective hydroxyl derivatives i.e. intermediates 3 and 4, we used pyridine·HCl as demethylating reagent and the products were isolated in quantitative yield. The keto-acid intermediates A and B (Scheme S1†) were synthesized by reacting phthalic anhydride with 3-(diethylamino)phenol and 3-(diphenylamino)phenol respectively. The DEMAP keto-acid intermediate 5 (Scheme S1†) was formed just after refluxing both the starting materials in toluene for 2 h, while Friedel–Craft reaction condition was followed for the synthesis of triphenylamine keto-acid intermediate 6 (Scheme S1†). Bis-hydroxyl intermediate 3 was treated with the both DEMAP and triphenylamine keto-acids i.e. intermediate 5 and 6 while tris-hydroxyl intermediate 4 was treated only with DEMAP keto acid i.e. intermediate 5 in TFA to acquire the desired products RH-1 to RH-5. Due to the presence of two and three reactive sites available on bis-hydroxyl intermediates 3 and tris-hydroxyl intermediates 4 respectively, we could isolate RH-1, RH-2, and RH-3 in the pure form by reacting intermediate 3 with both the keto-acids (intermediate A and B). While RH-4 and RH-5 were isolated by reacting DEMAP keto-acid (intermediate A) with tris-hydroxyl intermediates 4. The novel rhodamine derivates RH-1 to RH-5 (Fig. 1) were further confirmed by 1H and 13C NMR (Fig. 2).
 |
| | Fig. 1 Structures of the synthesized dyes RH-1 to RH-5 and dye RH-B for their open form. | |
 |
| | Fig. 2 Photographs of dyes RH-B and RH-1 to RH-5 in day and UV light for their open form in chloroform solvent. | |
Experimental section
Materials and methods
All the required chemicals were obtained from commercial sources and used as received without any further purification. The solvents used for synthesis and analytical measurements were obtained from the local suppliers, dried by following standard procedures and distilled prior to use. All the reactions were monitored by TLC (thin layer chromatography) with detection by UV light. Compound purification was obtained by using column chromatography, which was performed on 100–200 mesh silica as the stationary phase. 1H and 13C NMR spectral data were recorded on a Varian Cary Eclipse Australia 500 MHz instrument using TMS as an internal standard. The absorption spectra were recorded on PerkinElmer UV-visible spectrophotometer Lambda 25 and emission spectra were recorded on Varian Cary Eclipse fluorescence spectrophotometer using freshly prepared solutions at the concentration of 1 × 10−6 mol l−1 at room temperature using a 10 mm cuvette with a 5 (open form) and 10 nm (spirocyclic form) slit width. Fluorescence quantum yields were determined in different solvents by using Rhodamine 101 (Φ = 1 in methanol)55 as a reference standard using the comparative method. The absorption spectra of all the dyes as well as standard were measured at the absorbance below 0.1 to minimize the inner filter and re-absorption effect and the similar concentrations of solutions were used to calculate their emission intensity also. Absorption values were plotted against respective emission intensities and linear plots were obtained. Gradients for all dyes and standard were calculated. All the measurements were done by keeping the same slit width and their relative quantum yields were calculated by using eqn (1).| |
 | (1) |
where Φx = quantum yield of compound, ΦST = quantum yield of standard, Gradx = gradient of compound, GradST = gradient of standard, ηST2 = refractive index of solvent used for standard. ηx2 = refractive index of solvent used for compound.
Photophysical properties
The absorption and emission spectra of all the newly synthesized rhodamine derivatives were recorded in their spirocyclic as well as open form by using their 10 μM solutions in three different solvents i.e. toluene, chloroform and ethanol. The compounds were converted from their spirocyclic to open form by the addition of MeOH·HCl solution into it. The solvent was evaporated on rotavapour and the same procedure was repeated twice to get the fully open form of the rhodamine derivatives. Addition of organic solvents resulted in partial conversion of the salt form into the spirocyclic rhodamine derivatives. This is evident from the absorption peaks for both the open and spirocyclic forms in the photophysical studies. To acquire exclusively the open form of the rhodamine derivative requisite amount of trifluoroaceticacid (TFA) was added till the absorption peak of the spirocyclic derivative had completely disappeared. Fig. 3 and 4 represent normalized absorption and emissions spectra of all five dyes respectively for their spirocyclic as well as open form in chloroform solvent. Fig. S3 to S7† represents absorption and emission spectra dyes RH-1 to RH-5 respectively for their spirocyclic as well as open form in three different solvents (i.e. toluene, chloroform and ethanol) (Scheme 1).
 |
| | Fig. 3 Normalized absorption spectra of dyes RH-1 to RH-5 for their spirocyclic and open form in chloroform solvent (10−5 M concentrations) at room temperature. | |
 |
| | Fig. 4 Normalized emission spectra of dyes RH-1 to RH-5 for their spirocyclic and open form in chloroform solvent (10−5 M concentrations) at room temperature (λex: 322, 386, 307, 329 and 386 nm for RH-1 to RH-5 in cyclic form respectively and λex: 613, 576, 540, 627 and 603 nm for RH-1 to RH-5 in open form respectively). | |
 |
| | Scheme 1 Synthetic route for rhodamine RH-1 to RH-5. (a) 3-Bromo anisole, potassium-t-butoxide, 1,10 Phen/CuI, toluene (b) pyridine·HCl, 200 °C, overnight (c) intermediate A, TFA, 95 °C, 12 h (d) intermediate B, TFA, 95 °C, 12 h. (e) MeOH·HCl, reflux, 2 h. | |
From the structures of the synthesized dyes and combined absorption spectra of all the five dyes in their spirocyclic and open form (Fig. 3) they can be categorized into three different groups (i) plain bis and tris-DEMAP rhodamine derivatives (RH-2 and RH-5), (ii) hydroxyl-DEMAP rhodamine derivatives (RH-1 and RH-4) and (iii) triphenylamine rhodamine derivative (RH-3). Due to their structural similarity compound RH-1 and RH-4 show absorption λmax at 329 nm while compound RH-2 and RH-5 show absorption λmax at 386 nm in their spirocyclic forms. Compound RH-3 distinctly shows a very unique blue shifted absorption λmax at 307 nm mainly attributed to the π–π* transition from the extra triphenylamine phenyl rings. Similar patterns are observed for the open form, compound RH-3 shows blue shifted absorption λmax at 540 nm while compound RH-1 and RH-4 show red shifted absorption λmax at 613 and 627 nm respectively (Fig. 3). Compounds RH-2 and RH-5 show absorption λmax at 576 and 603 nm respectively in their open forms, shoulder peaks at 507 nm, 512 nm and 514 nm are observed for compound RH-1, RH-4 and RH-5 respectively in their open form.
The comparative emission spectra of all five rhodamine derivatives RH-1 to RH-5 are represented in Fig. 4 for their spirocyclic as well as open form. It is found that the dyes are more emissive in their open form in comparison to their respective spirocyclic form due to the increased conjugation length and effective charge transfer from donor to acceptor in their open form. Table 1 represents all photophysical parameters of dyes RH-1 to RH-5 for their spirocyclic as well as open form in chloroform solvent and Tables S1 to S5† represents photophysical parameters of dyes RH-1 to RH-5 respectively for their spirocyclic as well as open form in different solvents. In spirocyclic form the dyes emit in the range of 432 nm to 475 nm, while in the open form they emit from 619 nm to 719 nm. Similar to the spirocyclic form absorption spectra, the plain bis and tris-DEMAP rhodamine derivatives RH-2 and RH-5 show emission maxima in the same region (432–436 nm) while hydroxyl-DEMAP rhodamine derivatives RH-1 and RH-4 show emission in the similar region (471–475 nm) and in its spirocyclic form triphenylamine rhodamine derivative RH-3 shows very distinctive emission spectra at 436 nm. Particularly the hydroxyl derivatives show red shifted emission in their spirocyclic form. In their open form these compounds show very wide range of emission color from 619 nm (RH-2, pink color) to 719 nm (RH-3, blue color) in day as well as UV light. The remaining three compounds show emission at 630 nm (RH-5, red color), 657 nm (RH-1, violet color) and 660 nm (RH-4, faint blue color). Compound RH-5 shows maximum fluorescence intensity while compound RH-3 shows minimum fluorescence intensity in their spirocyclic as well as open form. It can be concluded from the normalized emission spectra of all rhodamine derivatives (Fig. 4), that the emission spectra for compound RH-2 is blue shifted while that of compound RH-3 is red shifted as compared to all other dyes in their open form. All these newly synthesized rhodamine derivatives show red shifted absorption as well as emissions than the reported Rhodamine B and 101 dyes, except compound RH-3 which show blue shifted absorption spectra than Rhodamine B and 101 (Table 2).
Table 1 Photophysical parameters of RH-1 to RH-5 in their spirocyclic and open form in chloroform
| Compounds |
λabs (nm) |
εmax × 104 (M−1 cm−1) |
fwhm (nm) |
λems (nm) |
Stokes shift |
f |
μeg (debye) |
Kr × 108 (cm−1) |
ΦF |
| (nm) |
(cm−1) |
| Cyclic form |
| RH-1 |
322 |
3.69 |
48 |
471 |
149 |
9824 |
0.73 |
7.08 |
2.99 |
— |
| RH-2 |
386 |
3.14 |
64 |
432 |
46 |
2759 |
0.63 |
7.18 |
3.62 |
— |
| RH-3 |
307 |
7.24 |
67 |
436 |
129 |
9638 |
1.87 |
11.06 |
9.52 |
— |
| RH-4 |
329 |
3.96 |
68 |
475 |
146 |
9343 |
0.92 |
8.02 |
4.41 |
— |
| RH-5 |
386 |
2.95 |
61 |
436 |
50 |
2971 |
0.55 |
6.73 |
2.98 |
— |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Open form |
| RH-1 |
613 |
10.04 |
80 |
660 |
47 |
1162 |
1.38 |
13.43 |
2.52 |
0.82 |
| RH-2 |
576 |
6.21 |
64 |
619 |
43 |
1206 |
0.65 |
8.90 |
1.95 |
0.54 |
| RH-3 |
540 |
9.48 |
100 |
719 |
179 |
4610 |
1.57 |
13.44 |
3.19 |
0.16 |
| RH-4 |
627 |
3.32 |
58 |
657 |
30 |
728 |
0.44 |
7.63 |
0.77 |
0.52 |
| RH-5 |
603 |
7.6 |
91 |
630 |
27 |
711 |
1.28 |
12.81 |
2.06 |
0.86 |
Table 2 Comparison between photophysical parameters of open form dyes RH-1 to RH-5 in acidic media verses RH-B and RH-101 in chloroform and 10% ethanol/water solution
| Solvent |
λabs (nm) |
λems (nm) |
Stokes shift (nm) |
| CHCl3 |
10% EtOH in H2O |
CHCl3 |
10% EtOH in H2O |
CHCl3 |
10% EtOH in H2O |
| RH-B |
557 |
— |
581 |
— |
24 |
— |
| RH-101 |
576 |
— |
603 |
— |
27 |
— |
| RH-1 |
613 |
616 |
660 |
664 |
47 |
48 |
| RH-2 |
576 |
566 |
619 |
647 |
43 |
81 |
| RH-3 |
540 |
573 |
719 |
708 |
179 |
135 |
| RH-4 |
627 |
615 |
657 |
659 |
30 |
44 |
| RH-5 |
603 |
608 |
630 |
656 |
27 |
48 |
The oscillator strength (f), and transition dipole moment (μeg) values for all the dye molecules RH-1 to RH-5 in different solvents are calculated using the expressions as per earlier reported procedures56 by utilizing the integrated absorption coefficient obtained from absorption spectra of their spirocyclic as well as open forms. Dye RH-3 due to its broad absorption spectra and high molar extinction coefficient shows higher values of oscillator strength (f) as well as transition dipole moment (μeg) both in their spirocyclic as well as open form, and higher values of molar extinction coefficient (εmax) were obtained for these dyes in their open form (Table 1). We also observed higher values of oscillator strength (f) and transition dipole moment (μeg) for them in their open form as compared to the respective spirocyclic form.
We calculated the values of radiative rate constant (Kr) for all these dyes in their spirocyclic as well as open form in different solvents by using Strickler Berg equation
| |
 | (2) |
where
![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) av
av is the average wave number corresponding to the 0–0 transition and the integral part is evaluated from the area under the curve of absorption and emissions spectra in different solvents and again we found higher values of
Kr for
RH-3 dye, both in spirocyclic as well as open form.
Effect of solvent on absorption and emission spectra of dyes RH-1 to RH-5 in their spirocyclic and open form. The photophysical studies of all the dyes were carried out in toluene, chloroform and ethanol. In general, the maximum values for molar extinction coefficient are observed in chloroform and those for minimum molar extinction coefficient values are in toluene, for both spirocyclic form and the open form of RH1–RH5 rhodamine derivative dyes (Fig. S1 to S5†).Similarly in the case of emission spectra, maximum emission intensities observed are in chloroform while minimum values are in ethanol in the open form (Fig. S1 to S5†). As we go from non-polar to polar solvent i.e. from toluene to ethanol, they are not showing any red shift in emission spectra for the open form suggesting that the dipole moment difference between the ground and excited state is minimum. In other words excited state of these dyes is not stabilized in polar environment. In the case of dyes RH-2 and RH-5 in their spirocyclic form, highly red shifted emission spectra are observed in toluene (Fig. S1 and S5†).
The emission spectra for the spirocyclic form of (Fig. S3 and S6†), dyes RH-1 and RH-4 i.e. compounds with extra hydroxyl group on triphenylamine phenyl ring are red shifted in chloroform and blue shifted in ethanol with increased emission intensity from ethanol to chloroform. Dye RH-3 emission spectra for the spirocyclic (Fig. S5†) form is red shifted in chloroform but with decreased emission intensity from toluene to chloroform. For all these dyes comparatively better variation in molar extinction coefficient as well as emission intensity is observed in their open form as compared to the spirocyclic form. No solvent polarity effect is observed on the spirocyclic form in absorption spectra for the dyes. It is conclusive from the calculated quantum yields for the dyes RH-1 to RH-5 in all the three solvents using Rhodamine 101 as a standard reference that for the open form the highest quantum yield are observed in chloroform and lowest one in ethanol, while the dye RH-1 and RH-5 shows highest quantum yield (0.82 and 0.86) and dye RH-3 shows the lowest quantum yield (0.16) in chloroform (Table 1).
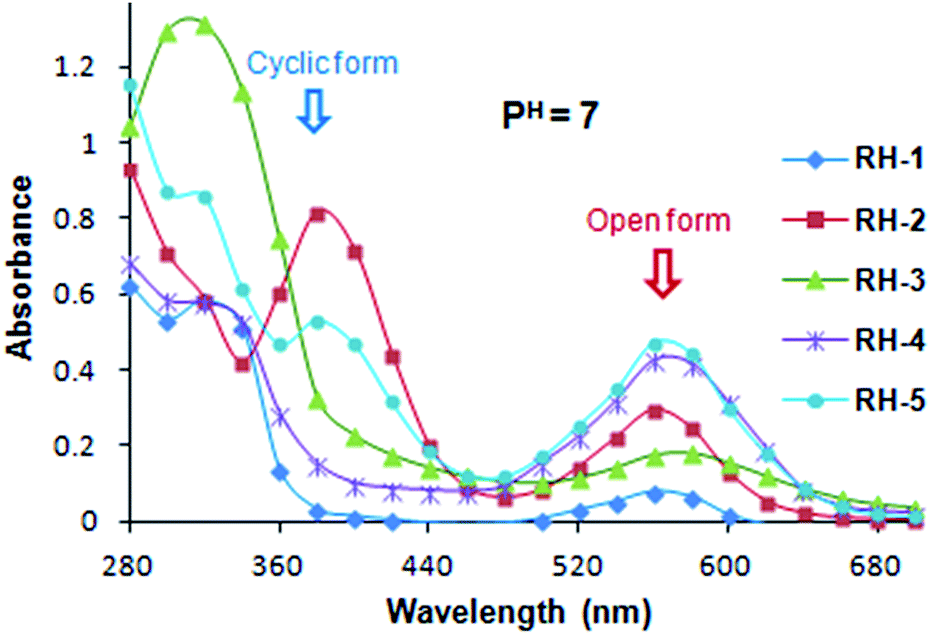
Absorption and emission spectra of dyes RH-1 to RH-5 in aqueous neutral (pH = 7) and acidic (pH = 1) medium. As the use of these dyes is mainly for their biological studies we evaluated the absorption and emission spectra of all dyes in aqueous media for their open form. The behaviour of their absorption and emission spectra in neutral as well as acidic media i.e. at pH = 7 and pH = 1 were studied. For this purpose we used 10 μM solutions of the dyes prepared in 10% ethanol–water system. The pH of the solution was adjusted using 0.1 N HCl and 0.1 N NaOH solutions. At neutral pH, all five dyes exist both in their spirocyclic as well as open form and shows comparatively higher molar extinction coefficient for their spirocyclic form than open form (Fig. 5). At neutral pH unlike the dyes RH-1 and RH-3 which prefer the spirocyclic form, high percentage of dyes RH-4 and RH-5 existed in open form. However the emission spectra for the dyes show negligible intensities at neutral pH.
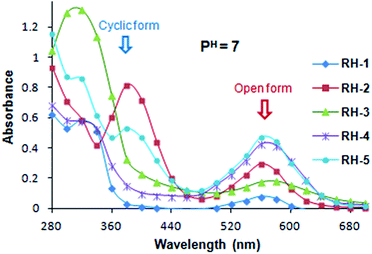 |
| | Fig. 5 Absorption spectra of RH-1 to RH-5 in 10% EtOH in water solutions at pH = 7 (3 × 10−5 M concentrations at room temperature). | |
The absorption and emission spectra (Fig. 6) for all five dyes and their normalized spectra at (pH = 1) acidic pH predominantly prefer open form with very good molar extinction coefficient (1.07 × 105 to 2.54 × 105 mol−1 cm−1). The hydroxyl substituted derivatives RH-1 and RH-4 show comparatively red shifted absorption λmax at 616 and 615 nm respectively, while plain rhodamine derivative RH-2 shows comparatively blue shifted absorption λmax as well as emission λmax at 566 and 646 nm respectively. Similarly another plain rhodamine derivative RH-5 shows comparative red shift absorption λmax (608 nm) but again blue shifted emission at 656 nm. Triphenylamine substituted derivative RH-3 shows very unique red shifted emission λmax at 705 nm. Detail absorption and emission values and respective Stokes of all five dyes in chloroform as well as in 10% ethanol in water are collected in Table 2.
 |
| | Fig. 6 Normalized absorption and emission spectra of RH-1 to RH-5 in 10% EtOH in water solutions at pH = 1 (3 × 10−5 M concentrations at room temperature and λex: 616, 566, 573, 615 and 608 nm for RH-1 to RH-5 in open form respectively). | |
Interconversion of spirocyclic to open form of dye RH-2 with the treatment of acid (TFA) in organic solvent. Conversion of spirocyclic ring form of rhodamine dyes to its open form is a well established phenomenon triggered by various metal ions and acids. The open form is highly conjugated exhibiting high molar extinction coefficient accompanied by red shifted absorption and emission peaks and increased emission intensities. In this context the present work discusses the effect of acid on the newly synthesized rhodamine dyes for the inter-conversion of spirocyclic to open form of dye RH-2 as a representative example. Requisite amount of trifluoroacetic acid (TFA) from 5 μl to 80 μl was added to a 10 μM solution of the spirocyclic form of the dyes in toluene till it was entirely converted into its opened form. In the absorption spectra with the increasing percentage of TFA in toluene, decreased absorbance of spirocyclic form at 385 nm with simultaneous increase in absorbance of its open form at 577 nm is observed as represented in Fig. 7; an isosbestic point of this inter-conversion is obtained at 438 nm. Similarly as represented in Fig. 8, in the emission spectra, with the increased percentage of TFA in toluene, resulted in the decreased emission intensity of spirocyclic form at 540 nm and simultaneous increase in emission intensity of its open form at 614 nm when excited at 385 nm, an isosbestic point for this conversion in emission spectra at 588 nm could be obtained. Interestingly when we excited at 577 nm, though the emission intensity of its open form increases with the increased percentage of TFA, it decreases in the presence of excess TFA; which may be due to the protonation of the donating nitrogen atom in the presence of excess TFA.
 |
| | Fig. 7 Absorption spectra of dye RH-2 with the increased percentage of TFA (5 to 80 μl) in toluene (10−5 M concentrations) at room temperature. | |
 |
| | Fig. 8 Emission spectra dye RH-2 with the increased percentage of TFA (5 to 80 μl) in toluene (10−5 M concentrations) at room temperature; (λex: 384 nm). | |
Study of viscosity sensitivity of rhodamine derivatives RH-1 to RH-5 in their open form. Triphenylamine based donor–acceptor compounds due to the rotating phenyl rings around C–N bond and C–C bond are found to show enhanced fluorescence in viscous medium and behave as fluorescent molecular rotors (FMR's). Recently Zhao et al. reported polarity based and viscosity sensitive emissions for the triphenylamine (TPA)-based rotors.53 Viscosity sensitivity of fluorescent organic compounds in NIR region is also useful for their biological applications. As all our triphenylamine based rhodamine derivatives emit in the deep red to NIR region in their open form we checked their viscosity sensitivity in the polar viscous mixture of solvents ethanol and glycerol in the presence of TFA to get their complete open form. It is observed that all the derivatives show high viscosity induced emission enhancement as represented in Fig. 9 and S8–S11.† Very high emission enhancement i.e. up to 24.56 fold is observed for dye RH-2 after the addition of 20 to 80% of glycerol in ethanol and hence this dye can be considered as fluorescent molecular rotor (FMR) emitting in the deep red region. With increasing percentage of glycerol in ethanol dyes RH-1 and RH-4 having extra hydroxyl group present on phenyl ring of triphenylamine shows comparatively less fluorescence enhancement (5.38 and 7.40 fold respectively) (Fig. 8 and S10†) and triphenylamine substituted derivative RH-3 shows 12.13 fold emission enhancement (Fig. S9†). Tri-substituted derivative RH-5 shows up to 8.43 fold of enhancement in fluorescence intensity after the addition of up to 80% of glycerol in ethanol. It is observed that as compared to the other derivatives dye RH-5 shows much linear fluorescence enhancement with the increased percentage of glycerol in ethanol. As we observed viscosity sensitivity of these compounds especially in their open form, the freely rotating phenyl ring in its open form attached to the xanthene core may be mainly responsible for the increased emission intensity with the increased percentage of glycerol in ethanol by hindering the rotation in viscous environment.
 |
| | Fig. 9 (a) Emission enhancement spectra of RH-1 in different ratio of glycerol (0 to 80%) in ethanol (0.5 × 10−5 M concentrations) at room temperature. (b) Plot of maximum emission intensity of RH-1 versus percentage glycerol (0 to 80%) in ethanol. | |
Conclusion
Five novel rhodamine derivatives purified by column chromatography in spirocyclic form were successfully isolated in moderate to good yields, which were converted into their open hydrochloride salt form by reacting with MeOH·HCl. All the dyes emit from deep red to NIR region and exhibit very good emission colors i.e. from pink to dark blue in day as well as UV light. The absorption and emissions for these dyes are comparatively red shifted unlike for the reported Rhodamine B and 101 compounds. The hydroxyl substituted rhodamine derivatives RH-1 and RH-4 shows red shifted emissions as compared to the plane rhodamine derivatives RH-2 and RH-5. Triphenylamine substituted rhodamine derivative RH-3 typically emits in the NIR region with very high Stokes shift of 135 nm. In their open form, all these rhodamine derivatives show very good fluorescence enhancement in viscous environment. Dye RH-2 particularly show very high fluorescence enhancement of 24.56 fold with the increased percentage of glycerol in ethanol. They also exhibited larger Stokes shift (44–135 nm) as compared to the small Stokes shift (25–30 nm) of Rhodamine B and 101.
Experimental section
3,3′-(Phenylazanediyl)diphenol 3
3-Methoxy-N-(3-methoxyphenyl)-N-phenyl aniline 1 (10 g, 32.74 mmol) was dissolved in 100 g of pyridine·HCl and heated to 200 °C for 10 h. After cooling to room temperature, water was added and solid precipitated out was filtered, dried and collected as crude product, which was further purified on column chromatography using 1% MeOH in DCM as the eluent to get the pure product. Yield = 7.1 g (78%); melting point = 156–158 °C. 1H NMR (500 MHz, CDCl3) δ 7.23–7.26 (m, 2H), 7.01–7.10 (m, 5H), 6.63 (dd, J = 8.0 and 2.0 Hz, 2H), 6.52 (t, J = 2.5 Hz, 2H), 6.47 (dd, J = 8.0 and 2.5 Hz, 2H), 4.87 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 156.2, 149.1, 147.3, 130.1, 129.2, 124.9, 123.3, 116.5, 110.8, 109.7. Elemental analysis calcd (%); Mol. formula: C18H15NO2 (C: 77.96, H: 5.45, N: 5.05, O: 11.54; found: C: 77.94, H: 5.39, N: 5.04, O: 11.55).
3,3′,3′′-Nitrilotriphenol 4
Tris(3-methoxyphenyl)amine 2 (15 g, 44.723 mmol) was dissolved in 150 g of pyridine·HCl and heated to 200 °C for 12 h. After cooling to room temperature, water was added and solid precipitated out was filtered, dried well and collected as crude product, which was further purified on column chromatography using 3% MeOH in DCM as the eluent to get the pure product. Yield = 9.7 g (74%); melting point = 265–268 °C. 1H NMR (500 MHz, DMSO-d6) δ 9.29 (s, 3H), 7.03 (t, J = 8.0 Hz, 3H), 6.36–6.39 (m, 9H). 13C NMR (126 MHz, DMSO-d6) δ 158.5, 148.8, 130.3, 115.1, 111.5, 110.7. Elemental analysis calcd (%); Mol. formula: C18H15NO3 (C: 73.71, H: 5.15, N: 4.78, O: 16.36; found: C: 73.72, H: 5.05, N: 4.72, O: 16.33).
3-(Diphenylamino)phenol 6
3-Methoxy-N,N-diphenylaniline 2 (16 g, 58.181 mmol) was dissolved in 80 g of pyridine·HCl and heated to 200 °C for 10 h. After cooling to room temperature, water was added and solid precipitated out was filtered, dried and collected as crude product, which was further purified on column chromatography using 10% EtOAc in hexane as the eluent to get the pure product. Yield = 12 g (79%); melting point = 98–100 °C. 1H NMR (500 MHz, CDCl3) δ 7.23–7.27 (m, 4H), 7.09–7.11 (m, 5H), 7.01–7.07 (m, 2H), 6.64 (dd, J = 8.5 and 2.0 Hz, 1H), 6.52 (t, J = 2.0 Hz, 1H), 6.46 (dd, J = 8.0 and 2.5 Hz, 1H), 4.60 (s, 1H). 13C NMR (126 MHz, CDCl3) δ 156.2, 149.3, 147.6, 130.1, 129.3, 124.7, 123.1, 116.1, 110.4, 109.4. Elemental analysis calcd (%); Mol. formula: C18H15NO (C: 82.73, H: 5.79, N: 5.36, O: 6.12; found: C: 82.72, H: 5.80, N: 5.29, O: 6.10).
2-(4-(Diethylamino)-2-hydroxybenzoyl)benzoic acid (intermediate A)
3-(Diethylamino)phenol (10 g, 60.52 mmol) and phthalic anhydride (9.85 g, 66.57 mmol) were added into anhydrous toluene (100 ml). The mixture was heated to reflux for 12 h and the solid precipitated out was filtered and purified by column chromatography (2% MeOH in chloroform). Yield = 11.6 g (61%); melting point = 202–205 °C. 1H NMR (500 MHz, CDCl3) δ 12.47 (s, 1H), 8.10 (d, J = 8.0 Hz, 1H), 7.63 (t, J = 7.5 Hz, 1H), 7.54 (t, J = 8.0 Hz, 1H), 7.36 (d, J = 7.5 Hz, 1H), 7.25 (s, 1H), 6.92 (d, J = 9.0 Hz, 1H), 6.27 (s, 1H), 6.19 (s, 1H), 3.38 (q, 4H), 1.19 (t, J = 7.0 Hz, 6H). Elemental analysis calcd (%); Mol. formula: C18H19NO4 (C: 68.99, H: 6.11, N: 4.47, O: 20.42; found: C: 68.97, H: 6.14, N: 4.39, O: 20.43).
2-(4-(Diphenylamino)-2-hydroxybenzoyl)benzoic acid (intermediate B)
3-(Diphenylamino)phenol 6 (0.5 g, 1.9 mmol), phthalic anhydride (0.35 g, 2.3 mmol), anhydrous aluminium chloride (0.38 g, 2.85 mmol) were mixed in ethylene dichloride (15 ml) under nitrogen atmosphere at 0 °C. The mixture was stirred at 0 °C for 1 h and then warmed to room temperature and maintained at room temperature for 2 h. After 2 h, reaction mass was stirred at 50 °C for 12 h and then it was cooled to room temperature, poured into 100 ml ice water with continuous stirring. A brownish yellow sticky precipitate was formed. The product was extracted with chloroform and the aqueous layer was separated. Organic layer was washed with water, dried using anhydrous Na2SO4 and then solvent was removed on rotatory evaporator. The product was purified by silica gel (100–200 mesh size) column chromatography to obtain greenish yellow solid as pure product. Yield: 0.4 g (55%); melting point = 188–192 °C. 1H NMR (500 MHz, CDCl3) δ 12.30 (s, 1H), 8.11 (d, J = 8.0 Hz, 1H), 7.63 (t, J = 7.5 Hz, 1H), 7.54 (t, J = 7.5 Hz, 1H), 7.36 (d, J = 7.5 Hz, 1H), 7.30–7.33 (m, 4H), 7.14–7.19 (m, 6H), 6.82–6.84 (d, J = 9.0 Hz, 1H), 6.43 (d, J = 2.0 Hz, 1H), 6.23 (dd, J = 9.0 and 2.5 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 199.4, 170.1, 164.6, 155.0, 145.4, 140.9, 133.7, 132.9, 131.1, 129.6, 129.4, 127.8, 127.4, 126.9, 125.5, 113.1, 110.0, 105.2. Elemental analysis calcd (%); Mol. formula: C26H19NO4 (C: 76.27, H: 4.68, N: 3.42, O: 15.63; found: C: 76.26, H: 4.64, N: 3.35, O: 15.65).
General procedure for the synthesis of RH-1 to RH-5
Initially we planned for the synthesis of RH-2 and RH-5 by treating DEMAP keto-acid intermediate A with bis-hydroxyl intermediate 3 and tris-hydroxyl intermediate 4 respectively, where we isolated mono and disubstituted RH-1 and RH-4 as by-products with the main products RH-2 and RH-5 in the reaction mixture. With RH-4 and RH-5, additional polar fluorescent spot was observed on TLC likely for the mono-substituted rhodamine derivative, attempts to isolate its pure form were unsuccessful. Similar strategy has been followed for the synthesis of RH-3, where one more mono-substituted product was observed on TLC, which we again failed isolate in its pure form. Di or tri-substituted triphenylamine hydroxyl derivative (1 eq.) and DEMAP or triphenylamine keto acid (2 eq. for RH-1, RH-2 and RH-3 and 3 eq. for RH-4 and RH-5) were dissolved in trifluoroacetic acid (10 ml) and stirred at 95 °C in a sealed tube for 12 h. After completion, the reaction mixture was poured in ice-water mixture and neutralized by saturated solution of NaHCO3. The solid precipitated out was filtered, dried and purified on column chromatography using 1–5% MeOH in chloroform as eluting system to get the pure products in their spirocyclic form.
3′-(Diethylamino)-6′-((3-hydroxyphenyl)(phenyl)amino)-3H-spiro[isobenzofuran-1,9′-xanthen]-3-one (RH-1). Compound 3 (0.3 g, 1.08 mmol) was reacted with intermediate A (0.84 g, 2.70 mmol). Yield = 0.2 g (36%); melting point = 286–291 °C. 1H NMR (500 MHz, CDCl3) δ 11.53 (s, 1H), 8.03 (d, J = 8.0 Hz, 1H), 7.72 (t, J = 7.5 Hz, 1H), 7.64 (t, J = 7.5 Hz, 1H), 7.58 (dd, J = 9.0 and 2.0 Hz, 1H), 7.40–7.43 (m, 3H), 7.34–7.19 (m, 6H), 7.06 (d, J = 2.0 Hz, 1H), 6.84 (d, J = 8.0 Hz, 1H), 6.74 (d, J = 9.0 Hz, 1H), 6.63 (s, 1H), 6.51 (dd, J = 9.0 and 2.0 Hz, 1H), 6.44 (d, J = 2.5 Hz, 1H), 3.36–3.37 (q, 4H), 1.17 (t, J = 7.0 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ 169.1, 152.5, 152.2, 149.6, 148.8, 145.9, 135.9, 130.5, 129.7, 129.6, 126.9, 126.8, 126.0, 125.1, 124.6, 118.7, 113.8, 110.1, 109.1, 104.8, 97.1, 83.8, 44.2, 12.7.Elemental analysis calcd (%); Mol. formula: C36H30N2O4 (C: 77.96, H: 5.45, N: 5.05, O: 11.54; found: C: 77.94, H: 5.36, N: 5.07, O: 11.49).
3′-(Diethylamino)-6′-(3′-(diethylamino)-3H-spiro[isobenzofuran-1,9′-xanthen]-3-one(phenyl)amino)-3H-spiro[isobenzofuran-1,9′-xanthen]-3-one (RH-2). Compound 3 (0.1 g, 1.08 mmol) was reacted with intermediate A (0.84 g, 2.70 mmol). Yield = 0.3 g (39%); melting point = 209–213 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.97 (d, J = 8.0 Hz, 2H), 7.77 (t, J = 7.5 Hz, 2H), 7.69 (t, J = 7.5 Hz, 2H), 7.39 (t, J = 7.0 Hz, 2H), 7.29 (d, J = 8.0 Hz, 2H), 7.16–7.23 (m, 3H), 6.73–6.77 (m, 4H), 6.63 (d, J = 8.5 Hz, 2H), 6.40–6.45 (m, 6H), 3.29 (q, 8H), 1.03 (t, J = 7.0 Hz, 12H). 13C NMR δ 169.1, 152.5, 152.2, 149.6, 148.8, 145.9, 135.9, 130.5, 129.4, 129.1, 127.1, 126.8, 126.2, 125.1, 124.5, 118.7, 113.8, 110.1, 109.1, 104.8, 97.3, 83.8, 44.1, 12.7. Elemental analysis calcd (%); Mol. formula: C54H45N3O6 (C: 77.96, H: 5.45, N: 5.05, O: 11.54; found: C: 77.97, H: 5.40, N: 5.07, O: 11.48).
3′-(Diethylamino)-6′-(3′-(diphenylamino)-3H-spiro[isobenzofuran-1,9′-xanthen]-3-one(phenyl)amino)-3H-spiro[isobenzofuran-1,9′-xanthen]-3-one (RH-3). Compound 3 (0.1 g, 0.36 mmol) was reacted with intermediate B (0.37 g, 0.90 mmol). Yield = 0.1 g (31%); melting point = 276–280 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.97 (d, J = 7.5 Hz, 2H), 7.78 (t, J = 7.5 Hz, 2H), 7.69 (t, J = 7.5 Hz, 2H), 7.27–7.34 (m, 13H), 7.03–7.11 (m, 18H), 6.55–6.59 (m, 8H). 13C NMR (126 MHz, DMSO-d6) δ 169.1, 152.5, 151.8, 150.1, 146.5, 136.1, 130.7, 130.3, 129.3, 126.5, 125.9, 125.2, 124.9, 124.5, 117.2, 111.5, 107.5, 82.9. Elemental analysis calcd (%); Mol. formula: C70H45N3O6 (C: 82.09, H: 4.43, N: 4.10, O: 9.37; found: C: 82.10, H: 4.38, N: 4.14, O: 9.33).
3′-(Diethylamino)-6′-((3-hydroxyphenyl)(3′-(diethylamino)-3H-spiro[isobenzofuran-1,9′-xanthen]-3-one)amino)-3H-spiro[isobenzofuran-1,9′-xanthen]-3-one (RH-4). Compound 4 (0.4 g, 1.36 mmol) was reacted with intermediate A (1.49 g, 4.77 mmol). Yield = 0.2 g (16%); melting point = >300 °C. 1H NMR (500 MHz, DMSO-d6) δ 9.59 (s, 1H), 7.97 (d, J = 7.5 Hz, 2H), 7.79 (t, J = 7.0 Hz, 2H), 7.71 (t, J = 7.0 Hz, 2H), 7.32 (d, J = 7.5 Hz, 2H), 6.75–6.80 (m, 4H), 6.54–6.65 (m, 5H), 6.41–6.44 (m, 7H), 3.29 (q, 8H), 1.04 (t, J = 7.0 Hz, 12H). 13C NMR (126 MHz, DMSO-d6) δ 169.2, 167.4, 165.1, 159.1, 153.9, 152.6, 152.2, 149.7, 148.8, 147.5, 140.5, 136.0, 134.6, 132.4, 131.2, 130.5, 130.3, 129.8, 129.4, 129.1, 128.1, 126.8, 125.1, 124.6, 118.9, 117.3, 113.7, 113.4, 113.2, 110.2, 109.7, 109.1, 104.8, 104.4, 97.4, 96.7, 83.8, 44.4, 12.7.Elemental analysis calcd (%); Mol. formula: C54H45N3O7 (C: 76.49, H: 5.35, N: 4.96, O: 13.21; found: C: 76.51, H: 5.29, N: 4.92, O: 13.23).
3′-(Diethylamino)-6′-[di(3′-(diethylamino)-3H-spiro[isobenzofuran-1,9′-xanthen]-3-one)]-3H-spiro[isobenzofuran-1,9′-xanthen]-3-one (RH-5). Compound 4 (0.4 g, 1.36 mmol) was reacted with intermediate A (1.49 g, 4.77 mmol). Yield = 0.2 g (12%); melting point = 225–229 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.99 (d, J = 7.5 Hz, 3H), 7.79 (t, J = 7.5 Hz, 3H), 7.72 (t, J = 7.5 Hz, 3H), 7.62 (d, J = 9.0 Hz, 1H), 7.35 (dd, J = 8.0 and 2.0 Hz, 3H), 7.16 (s, 3H), 6.99 (d, J = 8.5 Hz, 3H), 6.80 (d, J = 8.5 Hz, 3H), 6.54–6.42 (m, 8H), 3.31 (q, 12H), 1.05 (t, J = 7.0 Hz, 18H). 13C NMR (126 MHz, DMSO-d6) δ 169.1, 163.1, 154.4, 152.3, 149.7, 136.1, 130.7, 130.1, 129.1, 126.7, 125.2, 124.5, 122.3, 117.3, 114.7, 110.6, 109.3, 104.6, 97.3, 83.4, 44.2, 12.7.Elemental analysis calcd (%); Mol. formula: C72H60N4O9 (C: 76.85, H: 5.37, N: 4.98, O: 12.80; found: C: 76.81, H: 5.39, N: 5.02, O: 12.76).
Acknowledgements
Author SSK is thankful for University Grants Commission (UGC), India for a Research Fellowship. The authors acknowledge the comments from anonymous reviewers to improve the manuscript.
References
- B. Valeur, in Molecular Fluorescence, Wiley-VCH Verlag GmbH, 2001, pp. 3–19 Search PubMed
 .
. - E. Noelting and K. Dziewoński, Ber. Dtsch. Chem. Ges., 1905, 38, 3516–3527 CrossRef CAS .
- V. Dujols, F. Ford and A. W. Czarnik, J. Am. Chem. Soc., 1997, 119, 7386–7387 CrossRef CAS .
- J. Y. Kwon, Y. J. Jang, Y. J. Lee, K. M. Kim, M. S. Seo, W. Nam and J. Yoon, J. Am. Chem. Soc., 2005, 127, 10107–10111 CrossRef CAS PubMed .
- Y.-K. Yang, K.-J. Yook and J. Tae, J. Am. Chem. Soc., 2005, 127, 16760–16761 CrossRef CAS PubMed .
- M. H. Lee, J.-S. Wu, J. W. Lee, J. H. Jung and J. S. Kim, Org. Lett., 2007, 9, 2501–2504 CrossRef CAS PubMed .
- S.-K. Ko, Y.-K. Yang, J. Tae and I. Shin, J. Am. Chem. Soc., 2006, 128, 14150–14155 CrossRef CAS PubMed .
- H. Zheng, Z.-H. Qian, L. Xu, F.-F. Yuan, L.-D. Lan and J.-G. Xu, Org. Lett., 2006, 8, 859–861 CrossRef CAS PubMed .
- Y. Xiang, A. Tong, P. Jin and Y. Ju, Org. Lett., 2006, 8, 2863–2866 CrossRef CAS PubMed .
- X. Zhang, Y. Shiraishi and T. Hirai, Org. Lett., 2007, 9, 5039–5042 CrossRef CAS PubMed .
- Y. Xiang and A. Tong, Org. Lett., 2006, 8, 1549–1552 CrossRef CAS PubMed .
- S. Bae and J. Tae, Tetrahedron Lett., 2007, 48, 5389–5392 CrossRef CAS .
- X. Zhang, Y. Shiraishi and T. Hirai, Tetrahedron Lett., 2007, 48, 5455–5459 CrossRef CAS .
- X. Chen, T. Pradhan, F. Wang, J. S. Kim and J. Yoon, Chem. Rev., 2012, 112, 1910–1956 CrossRef CAS PubMed .
- K. H. Drexhage, in Dye Lasers, ed. F. P. Schafer, Springer, 1977 Search PubMed .
- K. H. Drexhage, Laser Focus, 1973, 9, 35–39 Search PubMed .
- W. Liu, M. Howarth, A. B. Greytak, Y. Zheng, D. G. Nocera, A. Y. Ting and M. G. Bawendi, J. Am. Chem. Soc., 2008, 130, 1274–1284 CrossRef CAS PubMed .
- C. Hara, K. Tateyama, N. Akamatsu, H. Imabayashi, K. Karaki, N. Nomura, H. Okano and A. Miyawaki, Brain Cell Biol., 2006, 35, 229–237 CrossRef CAS PubMed .
- G. A. Crosby and J. N. Demas, J. Phys. Chem., 1971, 75, 991–1024 CrossRef CAS .
- M. Bossi, V. Belov, S. Polyakova and S. W. Hell, Angew. Chem., Int. Ed., 2006, 45, 7462–7465 CrossRef CAS PubMed .
- F. Amat-Guerri, A. Costela, J. M. Figuera, F. Florido and R. Sastre, Chem. Phys. Lett., 1993, 209, 352–356 CrossRef CAS .
- Z. Zhou, M. Yu, H. Yang, K. Huang, F. Li, T. Yi and C. Huang, Chem. Commun., 2008, 3387–3389 RSC .
- M. H. Lee, H. J. Kim, S. Yoon, N. Park and J. S. Kim, Org. Lett., 2008, 10, 213–216 CrossRef CAS PubMed .
- M. Suresh, S. Mishra, S. K. Mishra, E. Suresh, A. K. Mandal, A. Shrivastav and A. Das, Org. Lett., 2009, 11, 2740–2743 CrossRef CAS PubMed .
- L. Long, W. Lin, B. Chen, W. Gao and L. Yuan, Chem. Commun., 2011, 47, 893–895 RSC .
- L. Yuan, W. Lin, Z. Cao, J. Wang and B. Chen, Chemistry, 2012, 18, 1247–1255 CrossRef CAS PubMed .
- L. Yuan, W. Lin, B. Chen and Y. Xie, Org. Lett., 2012, 14, 432–435 CrossRef CAS PubMed .
- L. Yuan, W. Lin, Y. Xie, B. Chen and J. Song, Chem.–Eur. J., 2012, 18, 2700–2706 CrossRef CAS PubMed .
- L. Yuan, W. Lin, Y. Xie, B. Chen and S. Zhu, J. Am. Chem. Soc., 2012, 134, 1305–1315 CrossRef CAS PubMed .
- X. Zhang, Y. Xiao and X. Qian, Angew. Chem., Int. Ed., 2008, 47, 8025–8029 CrossRef CAS PubMed .
- L. Yuan, W. Lin, K. Zheng and S. Zhu, Acc. Chem. Res., 2013, 46, 1462–1473 CrossRef CAS PubMed .
- W. Lin, L. Yuan, Z. Cao, Y. Feng and J. Song, Angew. Chem., Int. Ed., 2010, 49, 375–379 CrossRef CAS PubMed .
- M. Kumar, N. Kumar, V. Bhalla, H. Singh, P. R. Sharma and T. Kaur, Org. Lett., 2011, 13, 1422–1425 CrossRef CAS PubMed .
- V. Bhalla, R. M. Kumar, P. R. Sharma and T. Kaur, Inorg. Chem., 2012, 51, 2150–2156 CrossRef CAS PubMed .
- G.-S. Jiao, L. H. Thoresen and K. Burgess, J. Am. Chem. Soc., 2003, 125, 14668–14669 CrossRef CAS PubMed .
- R. Bandichhor, A. D. Petrescu, A. Vespa, A. B. Kier, F. Schroeder and K. Burgess, J. Am. Chem. Soc., 2006, 128, 10688–10689 CrossRef CAS PubMed .
- J. Han, J. Jose, E. Mei and K. Burgess, Angew. Chem., Int. Ed., 2007, 46, 1684–1687 CrossRef CAS PubMed .
- J. Fan, M. Hu, P. Zhan and X. Peng, Chem. Soc. Rev., 2013, 42, 29–43 RSC .
- Y. Q. Sun, J. Liu, X. Lv, Y. Liu, Y. Zhao and W. Guo, Angew. Chem., Int. Ed., 2012, 51, 7634–7636 CrossRef CAS PubMed .
- M. Fu, Y. Xiao, X. Qian, D. Zhao and Y. Xu, Chem. Commun., 2008, 1780–1782 RSC .
- Y. Koide, Y. Urano, K. Hanaoka, T. Terai and T. Nagano, J. Am. Chem. Soc., 2011, 133, 5680–5682 CrossRef CAS PubMed .
- Y. Koide, Y. Urano, K. Hanaoka, W. Piao, M. Kusakabe, N. Saito, T. Terai, T. Okabe and T. Nagano, J. Am. Chem. Soc., 2012, 134, 5029–5031 CrossRef CAS PubMed .
- Y. Koide, Y. Urano, K. Hanaoka, T. Terai and T. Nagano, ACS Chem. Biol., 2011, 6, 600–608 CrossRef CAS PubMed .
- M. Beija, C. A. M. Afonso and J. M. G. Martinho, Chem. Soc. Rev., 2009, 38, 2410–2433 RSC .
- Z. Tian, B. Tian and J. Zhang, Dyes Pigm., 2013, 99, 1132–1136 CrossRef CAS .
- T. Nguyen and M. B. Francis, Org. Lett., 2003, 5, 3245–3248 CrossRef CAS PubMed .
- J. Liu, Z. Diwu, W.-Y. Leung, Y. Lu, B. Patch and R. P. Haugland, Tetrahedron Lett., 2003, 44, 4355–4359 CrossRef CAS .
- E. David, J. Lejeune, S. Pellet-Rostaing, J. Schulz, M. Lemaire, J. Chauvin and A. Deronzier, Tetrahedron Lett., 2008, 49, 1860–1864 CrossRef CAS .
- M. Zhang, S. Zheng, X. Liu, Y. Long and B. Yang, Dyes Pigm., 2016, 125, 220–228 CrossRef CAS .
- M. Haidekker and E. Theodorakis, Org. Biomol. Chem., 2007, 5, 1669–1678 CAS .
- J. R. Lakowicz and B. R. Masters, J. Biomed. Opt., 2008, 13, 029901 CrossRef .
- A. Jacobson, A. Petric, D. Hogenkamp, A. Sinur and J. R. Barrio, J. Am. Chem. Soc., 1996, 118, 5572–5579 CrossRef CAS .
- F. Zhou, J. Shao, Y. Yang, J. Zhao, H. Guo, X. Li, S. Ji and Z. Zhang, Eur. J. Org. Chem., 2011, 2011, 4773–4787 CrossRef CAS .
- S. Kothavale and N. Sekar, Dyes Pigm., 2017, 136, 116–130 CrossRef CAS .
- T. Karstens and K. Kobs, J. Phys. Chem., 1980, 84, 1871–1872 CrossRef CAS .
- S. Kothavale and N. Sekar, Dyes Pigm., 2017, 136, 31–45 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: Absorption–emission spectra in different solvents and 1H and 13C NMR spectra of all intermediates and final rhodamine dyes were included. Tables containing photophysical parameters in three different solvents in their spirocyclic and open form are provided. See DOI: 10.1039/c6ra19899f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*