
Silane-terminated polyurethane applied to a moisture-curable pressure-sensitive adhesive using triethoxysilane
Abstract
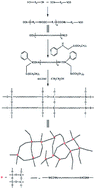
A silane-terminated polyurethane (SPU) modified by a silane end-capper anilinomethyltriethoxysilane (AMTES) was synthesized and acted as a pressure-sensitive adhesive (PSA) which could be moisture-cured at room temperature without releasing CO2. Tuning the NCO/OH ratio (R value) and polyols molecular weight altered the performances of the SPU, and satisfied different applications. Putting emphasis on the characteristic properties of PSA, the increase of R value enhanced the peel strength and holding power, but lowered the tack; increasing the polyols molecular weight reversed the trend of the peel strength, holding power and tack of PSA. The obtained PSA was thermostable at 250 °C and almost uncrystallized. Additionally, the glass transition temperature (Tg) and contact angle also changed with the R value and polyols molecular weight.
 Please wait while we load your content...
Please wait while we load your content...

