
Simple synthesis of lithium-doped sulfated titania nanoparticles and their high visible light photocatalytic activity under negative bias electrostatic field
Yu Guo*ab,
Junhua Chenb,
Zhijie Dingb,
Teng Guob,
Jumeng Weib,
Xiangju Yeb,
Weibing Xu*a and
Zhengfa Zhoua
aSchool of Chemistry and Chemical Engineering, Hefei University of Technology, Hefei, Anhui 230009, China. E-mail: weibingxu@hfut.edu.cn; guoyu4468@126.com; Fax: +86-0551-62901455; Tel: +86-0551-62901455
bCollege of Chemistry and Materials Engineering, Anhui Science and Technology University, Bengbu, Anhui 233100, China
First published on 12th October 2016
Abstract
Li-doped TiO2/SO42− nanoparticles was successfully synthesized via a simple calcinination process in a vacuum environment using Ti(SO4)2 and LiBr as precursors, and were characterized by TEM, XRD, IR, DLS, XPS and UV-vis (DRS). Li doping can reduce the diameters of TiO2 nanoparticles and affect the surface chemical forms and structures. The as-prepared photocatalysts with different LiBr contents exhibited a notably enhanced degradation efficiency for methylene blue, and exhibited the highest photocatalytic activity under UV light when the molar doping ratio of Li was 0.0125. Moreover, the most photocatalytically efficient sample also showed much higher activity for eliminating methylene blue under visible light (380 < λ < 700 nm) by applying a negative bias than P25 and the control samples without applying a negative bias. The introduced negative bias electrostatic fields could not only render TiO2 responsive to visible light, but was also able to increase the lifetime of the photo-excited charges in the doped semiconductor. This provides a facile, fast and universal method to rapidly degrade organic materials based on the Franz–Keldysh effect and the synergetic effects of the electrostatic force.
1. Introduction
Since 1972, titanium oxide (TiO2) has become the most representative oxide semiconductor material in fields such as solar energy conversion, electronics and electrochemistry owing to its excellent optical and electronic properties, chemical stability, biological inertness, non-toxicity and low cost.1–6 However, because of its wide band gap (anatase phase with a band gap of 3.2 eV and rutile phase with a band gap of 3.0 eV), TiO2 photocatalysts can only take enough effect when ultraviolet light is applied, which represents only a low percent (4–5%) of solar light.7–10 Thus, how to expand the absorption band of TiO2 photocatalysts to the visible light range is a key point in applying TiO2 materials as highly efficient photocatalysts. In addition, effectively controlling the recombination rate between photogenerated electron–hole pairs is also an issue which urgently needs to be solved. In addition, the specific optical, electric and catalytic properties, which arise from the intrinsic electronic structure, crystal phase, assembly, surface properties and particle size, should be investigated.11–14 Up to now, the preparation of improved TiO2 photocatalysts with excellent properties is still a great challenge.To achieve superior photocatalytic activity, various chemical methods, such as non-metal doping, metal doping and dye sensitization, have been developed to promote the photoresponse of TiO2 into the visible region.1,15–22 Recently, Li-doped TiO2 has been a research area of high current interest because of the focus towards the development of luminescent materials, new Li ion batteries and supercapacitors.23–27 Although discussions of the photocatalytic properties of Li-doped TiO2 are scarce, improved properties can be achieved by various methods, such as impregnation, solid grinding, high temperature diffusion synthesis and so on.28–36 On the other hand, an effective approach to improve the photocatalytic efficiency is further restraining the fast recombination of photogenerated electrons and holes (approximately 10 ns). Many efforts have proved that surface modification, such as increasing the number and the strength of surface acidic sites, and an externally applied bias can efficiently separate the electrons and holes.37–42 The strong acid sites on sulfated TiO2 increase the adsorption strength, which will result in an improved photocatalytic activity. Many researchers also reported that modification of the TiO2 catalyst’s surface with SO42− ions could efficiently enhance its photocatalytic activity due to the increase in the fraction of anatase, the surface area, and particularly the surface acidity.43–49 Compared to the photocatalytic process, the technique of photoelectrocatalytic oxidation for the degradation of the organic pollutants has attracted increasing attention to suppress the recombination of photogenerated electron–hole pairs by applying a small electric field.41,50 Many authors have also reported that photoelectrocatalysis is an efficient approach for the degradation of organic pollutants and photocatalytic disinfection under a certain bias but, unfortunately, there are some drawbacks to the photoelectrocatalytic technique being widely used.51–56 First, the requirement of using an electrolyte solution sets up a barrier to the mobile application in a standard three-electrode system; second, the electrochemical oxidation concomitantly takes place in the reaction, which hinders a clear understanding of the mechanism of the photocatalytic degradation.57
In this work, a simple pyrolysis method for preparing Li-doped TiO2 nanoparticles with surface sulfate is reported, in which the photocatalytic activities of four Li-doped TiO2/SO42− materials with different doping sequences under UV irradiation were compared side-by-side. To obtain the only anatase phase for the Li-doped samples, the maximum mol ratio of Li+ to Ti4+ was 0.04![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.58 Furthermore, a novel method to accelerate the photocatalytic reaction through an extra electric field was developed in this study, in which the photoelectrocatalytic performances of Li-doped TiO2/SO42− nanoparticles are dependent on the electrostatic forces produced by the external capacitor, instead of the DC power supply connected in series as in earlier reports. This will avoid electrochemical degradation effectively, even when the negative bias is more than the oxidation potential of the dyestuff. Through structure and composition analyses, the possible mechanisms were also elucidated for this enhancement of photoelectrocatalytic properties.
:1.58 Furthermore, a novel method to accelerate the photocatalytic reaction through an extra electric field was developed in this study, in which the photoelectrocatalytic performances of Li-doped TiO2/SO42− nanoparticles are dependent on the electrostatic forces produced by the external capacitor, instead of the DC power supply connected in series as in earlier reports. This will avoid electrochemical degradation effectively, even when the negative bias is more than the oxidation potential of the dyestuff. Through structure and composition analyses, the possible mechanisms were also elucidated for this enhancement of photoelectrocatalytic properties.
2. Experimental
2.1 Preparation of LST nanoparticles
Titanium sulfate (Ti(SO4)2, 96%), lithium bromide (LiBr, 99%), methylene blue (MB) and Degussa P25 were purchased from a commercial supplier (Sinopharm Chemical Reagent Co., Ltd.) and applied in our experiments without further purification. Deionized water was also used in this study.The Li-doped TiO2/SO42− nanoparticles were prepared by a calcination process using Ti(SO4)2 and LiBr as precursors. First, 6.0000 g of Ti(SO4)2 was dissolved in 50 mL of distilled water, and LiBr was added drop-wise into the above solution at room temperature. Four samples, with different Li/Ti molar ratios of 0.01, 0.0125, 0.02, and 0.04 were prepared and are labeled as, L0.01ST, L0.0125ST, L0.02ST and L0.04ST. Then, the mixture was dried at 100 °C until complete evaporation of the solvent and then calcined in a vacuum oven at 600 °C for 2 h to prepare the Li-doped TiO2/SO42− composites.
2.2 Sample characterization
The crystal phases of the P25 TiO2 and Li-doped SO42−/TiO2 photocatalysts were identified using XRD (D/Max-Ra, Rigaku, JP) with Cu Kα radiation (λ = 0.154 nm) in the range 2θ = 10–80° at a scan speed of 4° min−1. The average crystallite sizes of pure TiO2 and Li-doped TiO2 nanoparticles were determined from the anatase peak (101) using the Debye–Scherrer formula.59High resolution transmission electron microscopy (HR-TEM) images of typical samples were obtained using a 200 kV F20ST (FEI Company).
The size distributions of all samples were measured via dynamic light scattering (DLS) with a laser particle size analyzer (Bettersize Instruments, CN).
X-ray photoelectron spectroscopy (XPS) analysis was conducted on a VG ESCALB MK-II electron spectrometer using an Al Kα X-ray beam (1486.6 eV), and adventitious carbon (C 1s peak at 284.6 eV) was used to calibrate the binding energy.
The band gaps of the P25 and LST catalysts were determined using a UV-visible diffuse reflectance spectrophotometer (CARY 5000, Agilent Instruments, US) equipped with an integrating sphere and BaSO4 as reference. The UV-visible diffuse reflectance spectra were recorded at room temperature in the wavelength range of 200–800 nm, and the band gap energies were calculated according to eqn (1).
| Band gap (Eg) = hc/λ = 1240/λ | (1) |
Fourier transform infrared (FT-IR) spectra of the synthesized catalysts were recorded on a Thermo Nicolet-380 FT-IR spectrometer. The spectra were recorded in the range of 400–4000 cm−1 with the resolution of 4 cm−1 using KBr pellets.
2.3 Evaluation of the photocatalytic activity
All experiments are conducted at normal temperatures and pressures. Methylene blue (MB) was selected as the model chemical for evaluating the photocatalytic activity of the samples, and the initial pH was not adjusted. All samples were dispersed for 20 min using an ultrasound device in order to make them available for efficient surfactant adsorption. In order to select the conditions under which the photocatalytic performance was best, preliminary tests were performed for testing the photocatalytic activity of the prepared LST samples under direct UV irradiation.In a typical experiment, 0.1000 g of catalyst was dispersed in 100 mL of MB solution (20 mg L−1) in a small beaker inside a stainless steel container (Fig. 1). The mixtures were stirred constantly to avoid settling and ensure constant exposure of the photocatalyst to visible light radiation. The negative pole of the DC power supply was directly connected to the stainless steel sheathing; the positive pole was isolated to form the negative bias electrostatic field. A 500 W Xe lamp equipped with a UV cutoff filter was used as the source of visible light (intensity 4.56 × 104 Lux before optical filter, intensity 3.65 × 104 Lux after optical filter and a wavelength range of 380–700 nm). The light intensity of the Xe lamp is equivalent to the light intensity of the outdoor sunlight of Fengyang in Anhui province at 9:30 a.m. during April (intensity 4.61 × 104 Lux). Prior to irradiation, the suspension was maintained in the dark for 30 min in order to establish an adsorption–desorption equilibrium between the MB molecules and the catalyst. The absorbance of the MB aqueous solution was monitored over a time span of 80 min (at time intervals of 10 min) using a UV-vis spectrophotometer (UV-1800, Shimadzu Instruments, JP). After the dye was degraded under the constant potential mode, some aliquots of MB solution (10 mL) were taken out and separated by centrifugation, and then analyzed by monitoring the intensity variation of its main absorption peak (around 664 nm). The same procedure was performed for the P25 powder (the reference sample).
 | ||
| Fig. 1 Schematic diagram of experimental system for evaluating the photocatalytic activity. A, Xe lamp; B, optical filter; C, small beaker; D, stainless steel container; E, stirring bar; F, DC power source. | ||
According to the Beer–Lambert law, the absorbance of the solution is related to its solution’s concentration, that is, At = εlCt, where ε is the molar extinction coefficient, l is the light path length, and Ct is the solution concentration. For a reaction that follows a first-order kinetics, the degradation rate (η) and the apparent first-order constant (kapp) were calculated using eqn (2) and (3):
| η% (degradation rate) = (Aeq − At)/Aeq × 100% | (2) |
| ln(Aeq/At) = kappτ | (3) |
3. Results and discussion
3.1 Sample characterization
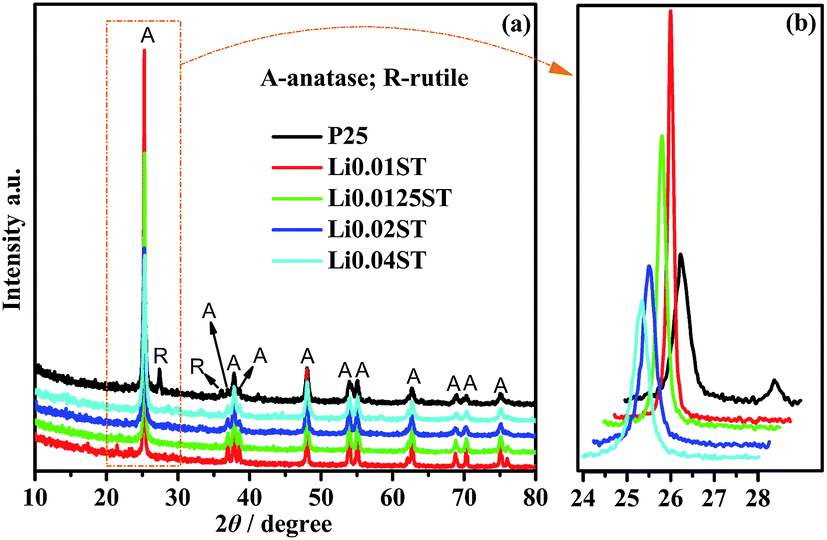 | ||
| Fig. 2 XRD patterns of the P25 and LST nanoparticles annealed at 600 °C. | ||
In previous work, Koudriachova et al. (2002) confirmed that with Li insertion in the lattice, the expansion of the lattice must lead to lengthening of the bond of Ti–O, which will accelerate the phase transformation from anatase to rutile,62 and many studies have proved that the effect of individual dopants on the structure of TiO2 is closely related to the preparative procedure of the materials. Bouattour et al. (2010) have shown that only the anatase phase is obtained for Li-doped TiO2 powder prepared by a solid grinding method at 400 °C. Nevertheless, a mixture of anatase and rutile phases is identified for Li-doped TiO2 synthesized by the sol–gel process using a mixture of acetic and hydrochloric acids as solvent in this work.32 When we analyze the LST samples prepared from the solid pyrolysis method, one can observe that anatase is the pure phase dominating the structure composition for samples with an increased Li/Ti molar ratio calcined at 600 °C. A similar conclusion has been drawn by López et al. (2010), who confirmed that only the anatase phase is obtained for a TiO2 sample doped with 1% Li+ and calcined at 400 °C.36 With respect to earlier reports that Li doping accelerates the phase formation of rutile, this interpretation is in disagreement with our study.32,62 The reason may be that most of the lithium ions exist in the particle surface rather than in the titanium oxide lattice in the process of thermal decomposition. The Ti–OH condensation reaction between TiO2 particles is greatly inhibited due to the formation of Ti–O–Li bonds, which contributes to the decreased particle size.
Fig. 3(a) and (b) give an overview of the typical TEM image of the L0.0125ST sample. It can be seen from Fig. 3(a) that L0.0125ST appears as spherical-like nanoparticles and aggregates. The HR-TEM image is shown in Fig. 3(b). It is clear that the crystal lattice scale of 0.35 nm in the darker section is in accordance with the (101) crystallographic plane of anatase TiO2. Fig. 3(c) shows that the particles’ sizes are fine enough and the average size is 20.82 nm. By comparison, it is found that the average size of the L0.0125ST particles obtained from the size distribution histogram analysis is fairly close to the result calculated by the Debye–Scherrer formula.
 | ||
| Fig. 3 (a) TEM image; (b) HR-TEM images; (c) the corresponding particle size distribution histogram of L0.0125ST and (d) size distribution plot for P25 and LST nanoparticles. | ||
Fig. 3(d) shows the particle size distribution for P25 and LST nanoparticle dispersions in deionized water. All samples were not dispersed using ultrasound beforehand in order to reflect the true size distribution of agglomerates at room temperature, and the agglomerate sizes were measured between 0.1 and 150 μm. The particle size distribution in Fig. 3(d) indicates a strongly decreased particle size as the dosage of LiBr increases in the synthesis process, which shows the maximum efficiency when the molar ratio of Li+ to Ti4+ is 0.04:1.63 All samples showed a multimodel size distribution, which indicated that particle aggregation had occurred. For instance, the particle size distribution of L0.0125ST was between 0.27 μm and 71.52 μm which is much higher than the individual particle size shown in Fig. 3(c). Thus, the particle size distribution of L0.0125ST in Fig. 3(d) actually corresponds to the sizes of agglomerates consisting of nanoparticles. Agglomerates with the average particle size of approximately 5.987 μm (P25), 25 μm (L0.01ST), 19.53 μm (L0.0125ST), 14.81 μm (L0.02ST) and 11.62 μm (L0.04ST) could be identified by the included software, demonstrating that doping TiO2 with Li inhibits the growth of the TiO2 particles. This variational trend is also consistent with the XRD results.
 | ||
| Fig. 4 (a) The UV-vis DRS spectra with band gap extrapolation lines for the P25, L0.01ST, L0.0125ST, L0.02ST and L0.04ST samples; (b) UV-vis diffuse reflectance spectra of LST and P25. | ||
| Sample | λabs (nm) | Eg (eV) |
|---|---|---|
| P25 | 397 | 3.12 |
| L0.01ST | 391.6 | 3.17 |
| L0.0125ST | 391.6 | 3.17 |
| L0.02ST | 391.6 | 3.17 |
| L0.04ST | 392.7 | 3.16 |
From the results above, it can be concluded that the tendency of the maximum absorption edge to decrease with decreasing particle size could be attributed to very small nanoparticles with quantum confinement effects. The deposition of lithium is a surface modification process rather than lattice doping.32,34 These results are in good agreement with the results of XRD and DLS. The different absorption features of L0.04ST compared with other LST samples can be attributed to the inward migration of lithium on the surface of the titanium, which may lead to interaction between the lithium ion and the titanium oxide lattice and the tendency of the band gap to decrease.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode or bending mode of the surface hydroxyl groups by comparison with other published results.43,59,67 Nevertheless, we found that Li doping seems to have an evident effect on the surface hydroxyl groups or the sulfur-containing functional groups from the FT-IR spectrum.
O stretching mode or bending mode of the surface hydroxyl groups by comparison with other published results.43,59,67 Nevertheless, we found that Li doping seems to have an evident effect on the surface hydroxyl groups or the sulfur-containing functional groups from the FT-IR spectrum.
 | ||
| Fig. 5 FT-IR spectra of (a) P25 and LST catalysts; (b) characteristic bands of SO42− bidentate ligand. | ||
Most obviously, it can be seen that there was a marked difference between the samples with the various lithium molar ratios added. The peak widths of the hydroxyl peaks in Li-doped TiO2 samples are notably broader than that of pure TiO2. The vibration at 1630 and 3430 cm−1 is very weak for L0.02ST and L0.04ST, revealing that Li-doped TiO2 nanoparticles had less hydroxyl groups on their surface. In photocatalytic reactions, the hydroxyl groups along with adsorbed water molecules play a crucial role as they react with photogenerated holes on the catalyst surface and yield hydroxyl radicals, which are potential oxidants for the degradation of pollutants.35,64 Thus, comparing various LST samples, L0.01ST and L0.0125ST can create more hydroxyl radicals, hence oxidising molecules adsorbed on the photocatalyst surface. Additionally, the 800–1500 cm−1 absorption bands of SO42− observed in L0.0125ST obviously decrease, widen and disappear, which means that the structure of SO42− was destroyed to a different extent because of the introduction of the lithium ion. Undoubtedly, the differences of the FT-IR spectra between all samples might be related to Ti–O–Li bond formation and the XRD results can also offer good evidence for the formation of the Ti–O–Li bond.
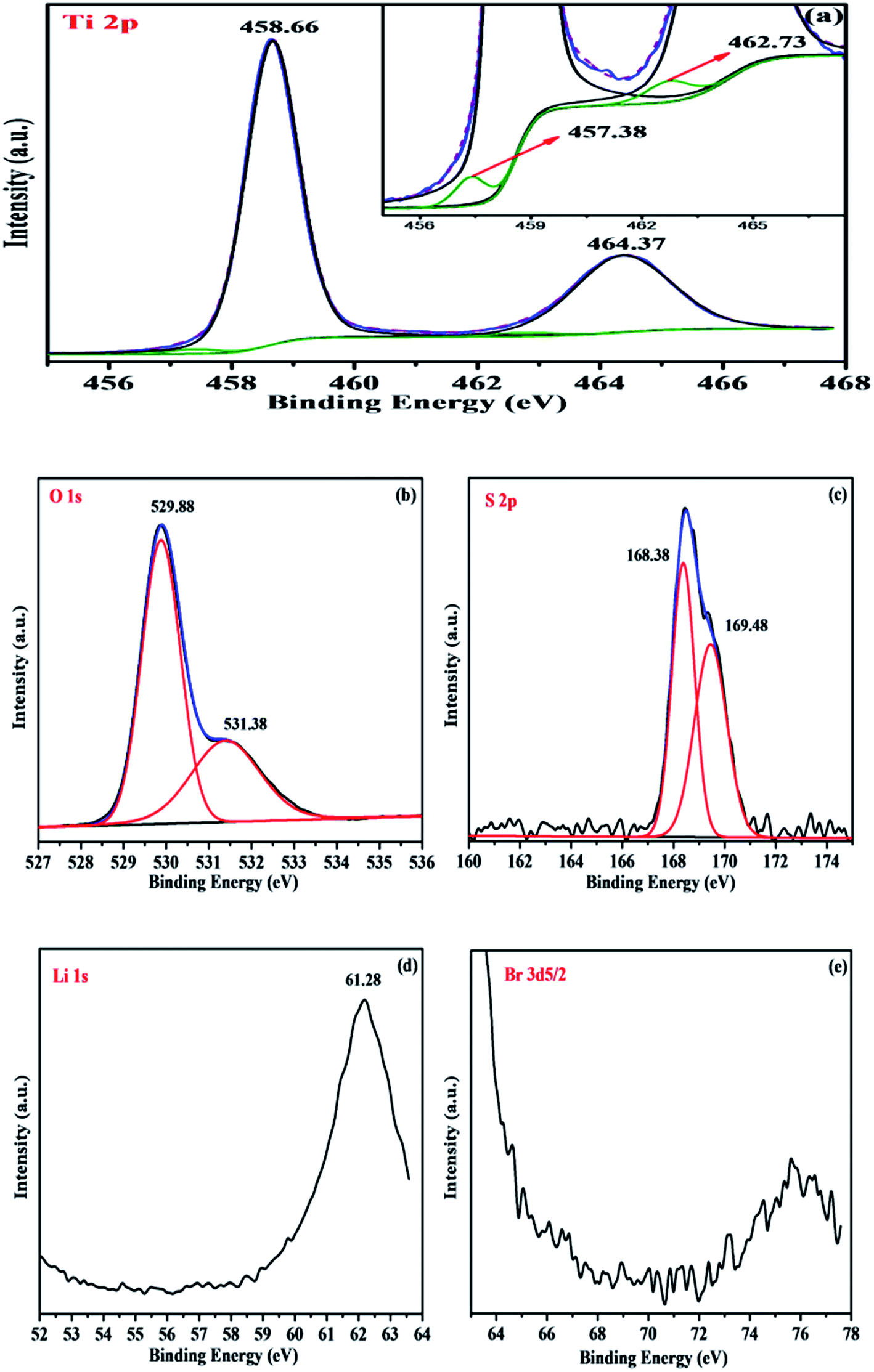 | ||
| Fig. 6 XPS of L0.0125ST nanoparticles: (a) the Ti4+ 2p orbital, (inset) the Ti3+ 2p orbital; (b) the O 1s orbital; (c) the S 2p orbital; (d) the Li 1s orbital; (e) the Br 3d orbital. | ||
The XPS pattern of O 1s of the L0.0125ST sample appears as asymmetric shapes which could be fitted with an intense component centred at 529.88 eV and a lower intensity peak centred at 531.38 eV (Fig. 6(b)). Comparing the values with those in the XPS standard manual, the binding energy of the 1s level of the O element in L0.0125ST at 529.88 eV is not strictly in accordance with the binding energy of anatase-TiO2 (O 1s at 529.2 eV). Thus, the first one was assigned to the presence of S–O–Ti and Li–O–Ti linkages, and the second one was ascribed to the hydroxyl oxygen (–OH) or chemisorbed water molecules on the surface of TiO2.38
Fig. 6(c) shows the binding energies at 168.38 and 169.48 eV, which were measured for S 2p3/2 and S 2p1/2, respectively. According to the values of the XPS standard manual and literature data, these binding energies are typical of elemental sulfur in the S6+ oxidation state. The peaks at 161–162.8 eV belonging to sulfide and those at 164 eV for elemental sulfur were not observed. The S6+ species might be present in the form of bidentate sulfate on the surface of TiO2, either chelating or bridging, as proposed in the literature.38,39,42,48 This is also consistent with the appearance of the band in the range 1500 to 900 cm−1 in the IR spectrum of the L0.0125ST sample.
From Fig. 6(d) and (e), such peaks centered at the binding energies (BE) of 56.6 (Li 1s) and 68.7 (Br 3d) eV cannot be found in the high resolution spectrum of Li 1s and Br 3d, which is due to the conjunction of four factors: low amount of LiBr (Li/Ti = 0.0125), ion loss during the calcination process, the position of the Li 1s peak (it is in the tail of Ti 3s with BE = 61 eV) and its very low sensitivity factor (0.02 compared to the one for Ti 3s equal to 0.16).32,33
Based on the results of IR and XRD, these features may be interpreted as the interaction between the sulfate anion and the titanium cation and homodispersion of trace Li ions in the particle surface.
3.2 Evaluation of photocatalytic activity
A series of measurements of the photocatalytic activity of each of the LST catalysts are used to rank them and select the one that performs best. 40 mg L−1 methylene blue (MB) was selected for the photocatalytic degradation under high pressure mercury lamp irradiation (300 W). Fig. 7 shows the photocatalytic activity of the P25 and LST samples for the UV adsorption driven MB degradation. From Fig. 7(a), it was seen that all samples exhibited superior degradation capacity for the methylene blue dye molecules under ultraviolet light irradiation. Among the LST catalysts, L0.0125ST exhibits the highest photocatalytic activity and removal percentage of MB, and can degrade more than 64% MB in 30 min. However, the photo-oxidation efficiencies for MB of P25 and the other LST samples (L0.01ST, L0.02ST, L0.04ST) were just 57.83%, 55.6%, 50.2% and 40.1%, respectively. It can also be seen that the overall photocatalytic performance of P25 is similar to that of L0.01ST. | ||
| Fig. 7 Comparison of photocatalytic properties of P25 and LST samples under UV. | ||
The kinetics of MB degradation with the P25 and LST samples are shown in Fig. 7(b). The line of best fit signifies the reaction rate constant, and hence the higher the slope, the faster the rate of the photocatalytic reaction. All the curves show that the photocatalytic degradation process by UV irradiation of the MB aqueous solution follows pseudo-first-order kinetics (Table 2). The values of the apparent rate constants (kapp) obtained from the slopes of the fitted lines in the second stage were 0.0134 min−1, 0.0149 min−1, 0.0177 min−1, 0.0102 min−1 and 0.0072 min−1 for P25, L0.01ST, L0.0125ST, L0.02ST and L0.04ST respectively. From the results of the evaluation of the performances of the LST samples, it is shown that L0.0125ST is an excellent photocatalyst for the photo-oxidation reaction.
| Sample | Pseudo-first-order kinetics | |
|---|---|---|
| kapp × 102 (min−1) | R2 | |
| P25 | 1.34 | 0.9607 |
| L0.01ST | 1.49 | 0.9717 |
| L0.0125ST | 1.77 | 0.9834 |
| L0.02ST | 1.02 | 0.9372 |
| L0.04ST | 0.72 | 0.9295 |
Generally, surface hydrophilicity and modification can affect the photocatalytic performance of TiO2. According to FT-IR, XRD and DLS results, it is easy to understand that Li doping will consume the OOH on the surface of TiO2 and photocatalysis experiments have also confirmed that the amount of surface hydroxyl groups is a key factor in affecting the photocatalytic performance. In addition, as a result of the high electronegativity of sulfur, the sulfate ion induces higher polarization than the P25 catalyst with a smaller particle size and a better capacity to be activated by visible light. The highly polarized state of the surface and the surface acidity would favor the trapping of electrons and enhance the photocatalytic activity. Predictably, for the P25 catalyst, the more photo-generated electrons and holes that are generated, the higher the recombination rate. This could also be a key reason for why the L0.0125ST sample with a completely sulfated structure exhibited the best photocatalytic performance among the P25 and LST samples, although the sulfating functional groups were also observed in other LST samples. Hence, the primary photocatalytic reactions of doped and undoped titania may be summarized as follows.70
| LS-TiO2/TiO2 + hν → e− + h+ | (4) |
| h+ + H2O → OH˙ + H+ | (5) |
h+ + ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) Ti–OH → OH˙ + H+ or h+ + Ti–O–Li → × Ti–OH → OH˙ + H+ or h+ + Ti–O–Li → ×
| (6) |
| e− + O2 → O2˙− | (7) |
| LS-TiO2/TiO2(OH˙), (O2˙−) + MB → decomposed products → CO2 + H2O | (8) |
The photocatalytic activity of the LST catalysts prepared according to the method we adopted may be closely related to the charge separation and the formation of OH˙ radicals. When the Li atomic ratio reached 0.02 and above in the TiO2 system, Li ions will consume too much OOH, thus decreasing the photocatalytic activity of TiO2. It should be obvious from Reaction (6) that the photogenerated holes can’t be trapped by the Ti–O–Li bonds. So we may reasonably conclude that the amount of the surface hydroxyl groups is the main reason for this photochemical performance difference.
To further investigate the photocatalytic properties, the photoelectrocatalytic efficiency of L0.0125ST and P25 were evaluated under visible light irradiation and a negative bias range from 0.0 to −7.5 V, and were monitored over 80 min of photoelectrocatalytic oxidation treatment. The degradation of MB without an external applied bias potential is illustrated as a reference.
Fig. 8 shows the photoelectrocatalytic kinetics of MB over P25 and the L0.0125ST samples, and the apparent first order rate constants, kapp, and the linear coefficient for the fitted line (Table 3) were calculated. As seen from Fig. 8(a) and (b), it is clear that the photo-oxidation efficiency of the P25 and L0.0125ST photocatalysts is strongly dependent on the negative bias and increases remarkably with the increasing negative bias. When no bias voltage is applied to the input, the photo-oxidation activity is the lowest, and the reduction ratio for MB is only 27.4% and 44.4% for 80 min illumination. Because the optical band gap of TiO2 is 3.12 and 3.17 eV which are selected for the present experimental conditions, it was hard to induce electron–hole carriers using visible light. Hence, it is very difficult to decompose MB without an external applied bias potential. By comparison, it is found that there are differences under visible light irradiation when the system was applied with −1.5 and −4.5 V bias potentials and the photoelectrocatalytic abilities of the L0.0125ST is obviously better. After 80 min, the final removal of MB was 94.5% (L0.0125ST −4.5 V), 64.9% (L0.0125ST −1.5 V), 45.4% (P25 −1.5 V), and 57.3% (P25 −4.5 V). Unexpectedly, when the value of the bias voltage is −7.5 V, the photoelectrocatalytic degradation for the P25 and L0.0125ST photocatalysts is almost identical and at their peak. The results clearly show that the enhanced photocatalytic activity cannot be completely attributed to the particle size and interfacial activity. It indicates that electrostatic forces could play a critical role in promoting the photocatalytic reaction. Under a cathodic bias electrostatic field, photogenerated charges can be separated effectively to carry out photocatalytic oxidation reactions, and the electron–hole recombination is inhibited due to the carrier lifetime being prolonged in the material. Therefore, the photodegradation process is improved, leading to the enhanced photocatalytic performance.
 | ||
| Fig. 8 Effect of visible light and different biases on the photoelectrocatalytic degradation of MB using P25 and L0.0125ST: (a) the extent of photoelectrocatalytic degradation of MB using P25; (b) the extent of photoelectrocatalytic degradation of MB using L0.0125ST; (c) the corresponding pseudo-first-order kinetic rate plot of P25; (d) the corresponding pseudo-first-order kinetic rate plot of L0.0125ST. | ||
| Sample | Pseudo-first-order kinetics | |
|---|---|---|
| kapp × 102 (min−1) | R2 | |
| P25 0 V | 0.41 | 0.9526 |
| P25 −1.5 V | 0.80 | 0.9881 |
| P25 −4.5 V | 1.08 | 0.9934 |
| P25 −7.5 V | 4.26 | 0.9683 |
| L0.0125ST 0 V | 0.76 | 0.9715 |
| L0.0125ST −1.5 V | 1.42 | 0.9756 |
| L0.0125ST −4.5 V | 3.39 | 0.9466 |
| L0.0125ST −7.5 V | 4.58 | 0.9498 |
Correspondingly, the photoelectrocatalytic kinetics of MB shows equally strong results, and the correlative coefficients (R2) of the linear regression function for the reaction kinetics curves are all higher than 0.9498. The apparent rate constants (kapp) followed the following trend: −7.5 V > −4.5 V > −1.5 V > 0 V (Fig. 8(c) and (d)). Interestingly, when applying a comparatively high negative bias for P25 and L0.0125ST, the photocatalytic reaction process is divided into two stages, and the value of the rate constant (kapp) will increase obviously in the latter reaction stage, in particular for L0.0125ST nanoparticles. Therefore, it is possible to infer that greater electrostatic interactions are more conducive to the photocatalytic degradation at low concentrations. This might be because more visible light is used to generate an excessive surplus of photoexcited carriers with the reduction of the MB concentration, eventually leading to a faster degradation reaction.
3.3 Mechanism for the photoelectrocatalytic activity under visible light irradiation
Based on the aforementioned experiments and analysis, the mechanisms for the photoelectrocatalytic reactivity are proposed. The high photoactivity of the as-prepared L0.0125ST may result from the following factors: (1) the grain size reaching nano-size, (2) the existence of –OH groups and sulfur groups on the surface, (3) the band gap narrowing under the electric field bias, and (4) the reduction of the photogenerated h+ and e− recombination rate due to the electrostatic interactions.For the L0.0125ST and P25 catalysts, the photoelectrocatalytic activities are improved by the electric field bias as shown in Fig. 9. We speculate that the mechanism might be as follows: when applying the electric field bias, a Franz–Keldysh effect happens. The externally applied potential can cause the bending of the band gap of TiO2, which is accomplished by a photon-induced tunnel effect. That means the band gap values decreased, and the intrinsic absorption edges of the TiO2 samples exhibited a red shift as the negative bias voltage increased (stage 1). For this, TiO2 and L0.0125ST can be activated by visible light. Simultaneously, there exists an electrostatic synergistic effect. Under the application of an additional electric field, the electron and hole move in opposite directions at different velocities, and form two relatively stable charge regions. Due to the electrostatic interactions of the electric field bias, the dynamic balance between carrier generation and recombination was destroyed, and the lifetimes of the carriers in the space charge region were prolonged by delaying their recombination. By contrast, the quantity of photoinduced electrons and holes on the surface of L0.0125ST and P25 will be significantly increased (stage 2). Subsequently, the excited electrons can be trapped by surface absorbed molecular oxygen (O2) to form superoxide anion radicals (O2˙−). Meanwhile, the photo-generated holes in the VB can be trapped by OH− or H2O species adsorbed on the catalyst surface to generate reactive hydroxyl radicals (OH˙) in aqueous media. These two radicals possessing powerful oxidizing abilities can degrade the MB completely into simpler molecules and corresponding minerals, which is responsible for the observed high photocatalytic activity of L0.0125ST and P25.71–76 In addition, the surface acidic sites and lithium ions of L0.0125ST are also believed to trap the photo-generated electrons, thus preventing the recombination of e− and h+.37,42,77 As a result, the L0.0125ST system exhibited higher photocatalytic activity for MB photodegradation than the P25 catalysts under 0 V, −1.5 V and −4.5 V supply. As the negative bias voltage increases to −7.5 V, electrostatic forces gradually become dominant, and directly determine the photocatalytic performance of the two catalysts.
 | ||
| Fig. 9 Schematic diagram of the photoelectrocatalytic mechanism of TiO2 under visible light irradiation. | ||
4. Conclusions
A series of LST photocatalysts with different Li/Ti molar ratios have been synthesized by a pyrolysis method using Ti(SO4)2 and LiBr as precursors. All samples show the polycrystalline anatase TiO2 phase and no rutile phase exists. The addition of lithium is not beneficial to prompt the phase transformation from anatase to rutile. TEM and DLS analyses show that Li-doping can reduce the diameters of the TiO2 nanoparticles and impart good water dispersibility without ultrasonic treatment. Ground state diffuse reflectance studies of LST reveal a tiny shift to the visible region, and the band gap values of the LST are almost unchanged, approximately 3.17 eV. The X-ray photoelectron spectroscopy (XPS) and FT-IR studies revealed that the doping elements did not enter the TiO2 crystal lattice to substitute Ti4+. Li+ is probably dispersed preferentially and uniformly at the surface of LST nanoparticles, whereas Li+ doping can also significantly affect the surface chemical forms and structures, e.g., the amount of hydroxyl groups decreases. The photoelectrocatalytic activity of the prepared catalysts under visible light irradiation was evaluated using MB as a pollutant model. The results showed a great enhancement in the photocatalytic efficiency with the application of different negative bias voltages. The introduced negative bias electrostatic fields could not only render TiO2 responsive to visible light, but were also able to increase the lifetime of the photo-excited charges in the doped semiconductor, consistent with a smaller recombination rate of the electron–hole pair. It is believed that this work may provide a new method for improving semiconductor photocatalysts.Acknowledgements
This work is supported by Anhui Science and Technology University Leading Academic Discipline Project (project number: AKZDXK2015A01).References
- S. Sood, S. K. Mehta, A. Umar and S. K. Kansal, New J. Chem., 2014, 38, 3127–3136 RSC.
- J. G. Yu, J. X. Low, W. Xiao, P. Zhou and M. Jaroniec, J. Am. Chem. Soc., 2014, 136, 8839–8842 CrossRef CAS PubMed.
- M. Răileanu, M. Crisan, I. Nitoi, A. Ianculescu, P. Oancea, D. Crisan and L. Todan, Water, Air, Soil Pollut., 2013, 224, 1548 CrossRef.
- X. Y. Pan, M. Q. Yang, X. Z. Fu, N. Zhang and Y. J. Xu, Nanoscale, 2013, 5, 3601–3614 RSC.
- N. Mir, K. Lee, I. Paramasivam and P. Schmuki, Chem.–Eur. J., 2012, 18, 11862 CrossRef CAS PubMed.
- A. Mukherji, P. Marschall, A. Tanksale, C. Sun, S. C. Smith, G. Q. Lu and L. Z. Wang, Adv. Funct. Mater., 2011, 21, 126–132 CrossRef CAS.
- T. Lin, C. Yang, Z. Wang, H. Yin, X. Lu, F. Huang, J. Lin, X. Xie and M. Jiang, Energy Environ. Sci., 2014, 7, 967–972 CAS.
- S. Kalathil, M. M. Khan, S. A. Ansari, J. Lee and M. H. Cho, Nanoscale, 2013, 5, 6323–6326 RSC.
- W. Lee, J. Lee, S. K. ochuveedu, T. Han, H. Jeong, M. Park, J. Yun, J. Kwon, K. No, D. Kim and S. Kim, ACS Nano, 2012, 6, 935–943 CrossRef CAS PubMed.
- C. W. Peng, T. Y. Ke, L. Brohan, M. Richard-Plouet, J. C. Huang, E. Puzenat, H. T. Chiu and C. Y. Lee, Chem. Mater., 2008, 20, 2426–2428 CrossRef CAS.
- Q. Y. Li, Y. Y. Xing, R. Li, L. L. Zong, X. D. Wang and J. J. Yang, RSC Adv., 2012, 2, 9781–9785 RSC.
- S. K. Das, M. K. Bhunia and A. Bhaumik, Dalton Trans., 2010, 39, 4382 RSC.
- (a) G. Liu, L. Z. Wang, H. G. Yang, H. M. Cheng and G. Q. Lu, J. Mater. Chem., 2010, 20, 831–843 RSC; (b) A. Fujishima, X. T. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- Y. Sakatani, D. Grosso, L. Nicole, C. Boissière, G. J. d. A. A. Soler-Illia and C. Sanchez, J. Mater. Chem., 2006, 16, 77 RSC.
- X. L. Jiang, X. L. Fu, L. Zhang, S. G. Meng and S. F. Chen, J. Mater. Chem. A, 2015, 3, 2271–2282 CAS.
- J. Liu, Q. C. Zhang, J. C. Yang, H. Y. Ma, M. O. Tade, S. B. Wang and J. Liu, Chem. Commun., 2014, 50, 13971–13974 RSC.
- S. Bingham and W. A. Daoud, J. Mater. Chem., 2011, 21, 2041–2050 RSC.
- E. Kowalska, O. O. Prieto-Mahaney, R. Abe and B. Ohtani, Phys. Chem. Chem. Phys., 2010, 12, 2344–2355 RSC.
- Z. Dai, G. Burgeth, F. Parrino and H. Kisch, J. Organomet. Chem., 2008, 694, 1049–1054 CrossRef.
- A. G. Agrios, K. A. Gray and E. Weitz, Langmuir, 2004, 20, 5911–5917 CrossRef CAS PubMed.
- Y. Bessekhouad, D. Robert and J. V. Weber, J. Photochem. Photobiol., A, 2004, 163, 569–580 CrossRef CAS.
- S. U. M. Khan, M. Al-Shahry and W. B. Ingler Jr, Science, 2002, 297, 2243–2245 CrossRef CAS PubMed.
- L. Kavan, J. Solid State Electrochem., 2014, 18, 2297–2306 CrossRef CAS.
- Y. J. Zhu, X. R. Zhao, L. B. Duan, X. J. Bai, H. N. Sun and W. F. Duan, J. Sol-Gel Sci. Technol., 2013, 67, 155–160 CrossRef CAS.
- C. W. Lai, S. Sreekantan, P. San E and W. Krengvirat, Electrochim. Acta, 2012, 77, 128–136 CrossRef CAS.
- V. Štengl, J. Velická, M. Maříková and T. M. Grygar, ACS Appl. Mater. Interfaces, 2011, 3, 4014–4023 Search PubMed.
- E. Baudrin, S. Cassaignon, M. Koelsch, J. P. Jolivet, L. Dupont and J. M. Tarascon, Electrochem. Commun., 2007, 9, 337–342 CrossRef CAS.
- M. A. Cortes-Jácome, M. Morales, C. A. Chavez, L. F. Ramírez-Verduzco, E. López-Salinas and J. A. Toledo-Antonio, Chem. Mater., 2007, 19, 6605–6614 CrossRef.
- W. C. Mackrodt, J. Solid State Chem., 1999, 142, 428–439 CrossRef CAS.
- A. Lewera, L. Timperman, A. Roguska and N. Alonso-Vante, J. Phys. Chem. C, 2011, 115, 20153–20159 CAS.
- Y. Bessekhouad, D. Robert, J. V. Weber and N. Chaoui, J. Photochem. Photobiol., A, 2004, 167, 49–57 CrossRef CAS.
- W. Kallel, S. Bouattoura, L. F. Vieira Ferreirab and A. M. Botelho do Regob, Mater. Chem. Phys., 2009, 114, 304–308 CrossRef CAS.
- S. Bouattour, A. M. Botelho do Rego and L. F. Vieira Ferreira, Mater. Res. Bull., 2010, 45, 818–825 CrossRef CAS.
- Z. Hamden, S. Boufi, D. S. Conceic, A. M. Ferraria, A. M. Botelho do Rego, D. P. Ferreira, L. F. Vieira Ferreira and S. Bouattour, Appl. Surf. Sci., 2014, 314, 910–918 CrossRef CAS.
- H. F. Jiang, H. Y. Song, Z. X. Zhou, X. Q. Liu and G. Y. Meng, J. Phys. Chem. Solids, 2007, 68, 1830–1835 CrossRef CAS.
- T. López, J. Hernandez-Ventura, R. Gómez, F. Tzompantzi, E. Sánchez, X. Bokhimi and A. Garcia, J. Mol. Catal. A: Chem., 2001, 101–107 CrossRef.
- C. H. Chen, Q. W. Liu, S. M. Gao, K. Li, H. Xu, Z. Z. Lou, B. B. Huang and Y. Dai, RSC Adv., 2014, 4, 12098–12104 RSC.
- P. Goswami and J. N. Ganguli, RSC Adv., 2013, 3, 8878–8888 RSC.
- R. Velmurugan, B. Krishnakumar and M. Swaminathan, Mater. Sci. Semicond. Process., 2014, 25, 163–172 CrossRef CAS.
- Y. Y. Qin, Y. L. Li, Z. Tian, Y. L. Wu and Y. P. Cui, Nanoscale Res. Lett., 2016, 11, 32 CrossRef PubMed.
- Y. Cui, H. Du and L. S. Wen, Environ. Chem. Lett., 2009, 7, 321–324 CrossRef CAS.
- X. C. Wang, J. C. Yu, P. Liu, X. X. Wang, W. Y. Su and X. Z. Fu, J. Photochem. Photobiol., A, 2006, 179, 339–347 CrossRef CAS.
- Z. C. Wang and H. F. Shui, J. Mol. Catal. A: Chem., 2007, 263, 20–25 CrossRef CAS.
- G. Colón, M. C. Hidalgo and J. A. Navío, Appl. Catal., B, 2003, 45, 39 CrossRef.
- W. R. Su, Y. L. Chen and X. Z. Fu, et al., Chem. Res. Chin. Univ., 2002, 23, 1398 CAS.
- D. S. Mugli and L. Ding, Appl. Catal., B, 2001, 32, 181 CrossRef.
- T. Ohno, M. Akiyoshi and T. Umebayashi, et al., Appl. Catal., A, 2004, 265, 115–123 CrossRef CAS.
- S. X. Liu and X. Y. Chen, J. Hazard. Mater., 2008, 152, 48–55 CrossRef CAS PubMed.
- Q. Sun, Y. C. Fu, J. W. Liu, A. Auroux and J. Y. Shen, Appl. Catal., A, 2008, 334, 26–34 CrossRef CAS.
- K. Vinodgopal, S. Hotchandani and P. V. Kamat, J. Phys. Chem., 1993, 97, 9040–9044 CrossRef CAS.
- P. A. Christensen, T. P. Curtis and T. A. Egerton, et al., Appl. Catal., B, 2003, 41, 371–386 CrossRef CAS.
- T. An, G. Li and X. Zhu, et al., Appl. Catal., A, 2005, 279, 247–256 CrossRef CAS.
- Y. Cui, H. Du and L. Wen, Environ. Chem. Lett., 2009, 7, 321–324 CrossRef CAS.
- H. Wu and Z. Zhang, J. Solid State Chem., 2011, 184, 3202–3207 CrossRef CAS.
- J. Shang, Y. Zhang and T. Zhu, et al., Appl. Catal., B, 2011, 102, 464–469 CrossRef CAS.
- X. Yu, X. Han and Z. Zhao, et al., Nano Energy, 2015, 11, 19–27 CrossRef CAS.
- J. Shang, S. Xie and T. Zhu, et al., Environ. Sci. Technol., 2007, 41, 7876–7880 CrossRef CAS PubMed.
- A. Henningsson, M. P. Andersson, P. Uvdal, H. Siegbahn and A. Sandell, Chem. Phys. Lett., 2002, 360, 85–90 CrossRef CAS.
- C. Chen, Q. Liu, S. Gao, K. Li, H. Xu and Z. Lou, et al., RSC Adv., 2014, 4, 12098–12104 RSC.
- M. Zhang, Y. Y. Xu, J. G. Lv, L. Yang, X. H. Jiang, G. He, X. P. Song and Z. Q. Sun, Nanoscale Res. Lett., 2014, 9, 636 CrossRef PubMed.
- M. Y. Li, L. Xiong, Y. Y. Chen, N. Zhang, Y. M. Zhang and H. Yin, Sci. China, Ser. B: Chem., 2005, 48, 297–304 CrossRef CAS.
- M. V. Koudriachova, N. M. Harrison and S. W. de Leeuw, Comput. Mater. Sci., 2002, 24, 235 CrossRef CAS.
- Y. Feng, J. N. Hart, R. J. Patterson and A. Lowe, Mater. Lett., 2015, 139, 31–34 CrossRef CAS.
- Z. X. Pei, L. Y. Ding, W. H. Feng, S. X. Weng and P. Liu, Phys. Chem. Chem. Phys., 2014, 16, 21876–21881 RSC.
- W. Ho, J. C. Yu and S. Lee, J. Solid State Chem., 2006, 179, 1171–1176 CrossRef CAS.
- C. Han, M. Pelaez, V. Likodimos, A. G. Kontos, P. Falaras, K. O’Shea and D. D. Dionysiou, Appl. Catal., B, 2011, 107, 77–87 CrossRef CAS.
- N. Sharotri and D. Sud, New J. Chem., 2015, 39, 2217–2223 RSC.
- H. Kamani, S. Nasseri, M. Khoobi, R. N. Nodehi and A. H. Mahvi, J. Environ. Health, 2016, 14, 3 Search PubMed.
- S. Liu and X. Chen, J. Hazard. Mater., 2008, 152, 48–55 CrossRef CAS PubMed.
- H. Jiang, H. Song and Z. Zhou, et al., J. Phys. Chem. Solids, 2007, 68, 1830–1835 CrossRef CAS.
- M. M. Momeni, Appl. Phys. A, 2015, 119, 1413–1422 CrossRef CAS.
- R. Kralchevska, M. Milanova, M. Tsvetkov, D. Dimitrov and D. Todorovsky, J. Mater. Sci., 2012, 47, 4936–4945 CrossRef CAS.
- S. Sood, S. K. Mehta, A. Umar and S. K. Kansal, New J. Chem., 2014, 38, 3127–3136 RSC.
- S. J. Yuan, X. W. Li and X. H. Dai, RSC Adv., 2014, 4, 61036–61044 RSC.
- M. M. Khan, S. A. Ansari, D. Pradhan, M. O. Ansari, D. H. Han, J. Leea and M. H. Cho, J. Mater. Chem. A, 2014, 2, 637–644 CAS.
- S. B. Rawal, S. Bera, D. Lee, D. J. Jang and W. I. Lee, Catal. Sci. Technol., 2013, 3, 1822–1830 CAS.
- S. Bouattor, W. Kallel, A. M. Botelho do Rego, L. F. Vieira Ferreira, I. F. Machado and S. Boufi, Appl. Organomet. Chem., 2010, 10, 692–699 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |