
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Soluble silicon patterns and templates: calcium phosphate nanocrystal deposition in collagen type 1†
G.
Birdi-Chouhan
a,
R. M.
Shelton
b,
J.
Bowen
 c,
P.
Goldberg-Oppenheimer
a,
S. J.
Page
d,
J. V.
Hanna
d,
A.
Peacock
e,
A. J.
Wright
e and
L. M.
Grover
*a
c,
P.
Goldberg-Oppenheimer
a,
S. J.
Page
d,
J. V.
Hanna
d,
A.
Peacock
e,
A. J.
Wright
e and
L. M.
Grover
*a
aSchool of Chemical Engineering, University of Birmingham, Birmingham, B15 2TT, UK. E-mail: l.m.grover@bham.ac.uk
bSchool of Dentistry, University of Birmingham, Birmingham, B4 6NN, UK
cDepartment of Engineering and Innovation, The Open University, Milton Keynes, MK7 6AA, UK
dDepartment of Physics, University of Warwick, Coventry, CV4 7AL, UK
eSchool of Chemistry, University of Birmingham, Birmingham, B15 2TT, UK
First published on 11th October 2016
Abstract
Patterned mineralisation is a feature of many hard-tissues. The impressive mechanical properties exhibited by such tissues can be, in part, attributed to the patterned deposition of mineral within the organic matrix. Although not thermodynamically favourable, the deposition of calcium phosphate based mineral within collagen fibres occurs in vivo in bone and dentine. As a consequence, numerous researchers have investigated how matrix proteins may be conditioned to enable patterned mineral deposition to recapitulate the structures found in nature. In this study, we have demonstrated that this patterned mineralisation of collagen type I may be induced simply by the pretreatment of the collagen with orthosilicic acid (OSA). The OSA treatment of the collagen resulted in a structural change to the collagen fibres, modifying the fibril diameter and changing the kinetics of fibre formation. NMR demonstrated that the OSA was preferentially located at the termini of the procollagen fibrils, thereby templating the formation of apatitic calcium phosphate crystals within the collagen fibrils (as shown using TEM, EDX and SAED). This work demonstrates how simple inorganic ions can have potent effects on structuring biological precipitates and suggests why trace quantities of silicon ions are essential to the formation of healthy hard tissues.
Introduction
The essential role of silicon in bone biology was first reported by Carlisle (1974) and Schwarz et al. (1972). They demonstrated that decreasing the levels of silicon to a critical level (<0.5 wt%) in chick1 and rat2 diets resulted in bone and cartilage deformations, whereby the bone did not ossify and so remained flexible. Silicon is present in vivo at 0.5 wt% within newly forming bone (osteoid). This association has led to speculation that soluble silicon, orthosilicic acid (OSA), may play a role during the early stages of mineralized tissue formation indirectly, through its interaction with connective tissues.3 Indeed, it was hypothesized that OSA interacts directly with the glycosaminoglycan network within these tissues, through the formation of silanoate (R–O–Si–O–R) cross-links that were critical to the stability of the maturing tissue.4 In the forty years since this hypothesis was made, there has been no further proof that demonstrates that such interactions exist or that they are essential to tissue structure. More recent work has focused exclusively on how soluble silicon influences the cells that are found in the newly forming tissue and has largely ignored its role in organizing and manipulating the structure of the extracellular matrix. Soluble silicon has been shown to stimulate human osteoblasts and osteoblast-like cells to secrete type I collagen and other bone markers related to osteoblast maturation and bone formation.5,6 Even though silicon has been shown to increase the osteogenic characteristics of osteoblasts, the precise role and localization of silicon remains unclear in bone biology. Numerous studies have also demonstrated that the substitution of silicon into biomaterials used for bone reconstruction can enhance bone formation in the site adjacent to the implant material.7–9 As a consequence, a number of materials are now commercially available that are designed to deliver silicon to the implant site in vivo.10Bone is a tissue that is structured at the nano-scale and comprises collagen fibrils into which calcium phosphate nanocrystals are deposited at 67 nm intervals termed hole zones.11 It is this nanoscale organisation that is responsible for the highly impressive toughness and strength of mineralised bone.12,13 Interestingly, however, collagen fibrils alone are unable to template patterning of such regular deposits of calcium phosphate. When collagen gels are left in a saturated solution of calcium and phosphate ions, mineralization will occur on the surface of the fibrils as opposed to within the fibrillar structure itself.14 The lack of deposition of calcium phosphate mineral within these “hole-zones” has been attributed to the fact that deposition of mineral within the collagen fibril is thermodynamically unfavourable.15 A number of non-collagenous proteins (NCPs) that could potentially overcome this thermodynamic barrier and stimulate intrafibrillar deposition16–18 have been identified. These NCPs have been shown to mediate mineral deposition within the collagen fibril and maintain the amorphous nature of the calcium phosphate initially during deposition onto the collagen fibril.16 This enhancement in the stability of the amorphous phase has been hypothesised to be one of the factors responsible for enabling intrafibrillar precipitation to take place.19 Attempting to mimic this process in vitro through the use of negatively charged small molecules such as poly(aspartic acid), generated intrafibrillar deposition through the formation of a polymer induced liquid precursor (PILP) that may have diffused into the fibril.20 More recently, it has been reported that simply making the collagen gels basic (pH 8.5) resulted in an increase in the anionic nature of the collagen and may cause local deamination of the collagen fibril. This in-turn generates a further local reduction in pH and may drive the deposition of the calcium phosphate nanocrystals within the collagen fibril.21 It is worth noting that all of these processes require the use of high concentrations of the component molecules to stimulate template mineralization. Although understanding of bone formation has improved, it remains unclear precisely how and why silicon species seem to be critical to the process. We hypothesise that the role of silicon in bone is not limited to stimulation of osteoblasts to secrete more collagen,5 but could be as a consequence of the patterning of the collagenous matrix by orthosilicic acid. We propose that soluble silicon induces patterned mineral deposition in collagen fibrils and serves as a precursor by preparing the collagen template for intrafibrillar apatite deposition.
Results and discussion
Initially, viability and proliferation studies were carried out on populations of MC-3T3-E1 cells cultured in the presence of 5 and 20 μg ml−1 of OSA. Cell viability was significantly (p < 0.05) higher when cells were cultured with 5 μg ml−1 OSA on day 7 and 10, when compared with untreated cells and reached 100% confluency by day 14. Cells cultured with 20 μg ml−1 showed a decrease in cell viability from day 7, this was considered as the maximum dose for MC-3T3-E1 cell survival (Fig. S1†). Furthermore bone nodule formation was evaluated using Alizarin Red-S staining. Following 14 days of culture in the absence of osteogenic mediators such as, ascorbic acid, β-glycerophosphate and dexamethasone, mineral was noted in all specimens (Fig. 1a). It was particularly noteworthy, that larger and more intensely stained mineral nodules were formed in the absence of OSA rather than in its presence. Indeed when the same threshold was applied, using Image-J to each sample using the most intense staining of the control specimen as 100, there appeared to be a larger area of mineral formation in the control sample as opposed to treated specimens (Fig. 1b). Superficially it appeared that the OSA inhibited bone nodule formation in these cell cultures. Quantitative analysis of the dissolved stain from the cultures, however, demonstrated that there was little difference in the quantity of mineral present in each specimen at each time point when normalized against total DNA content (Fig. 1c). It has previously been shown that in the presence of silicon, calcium phosphate (CaP) nodule formation is reduced on the surface of osteoblast cells, but instead mineral deposits are noted on the periphery and within the collagen matrix.22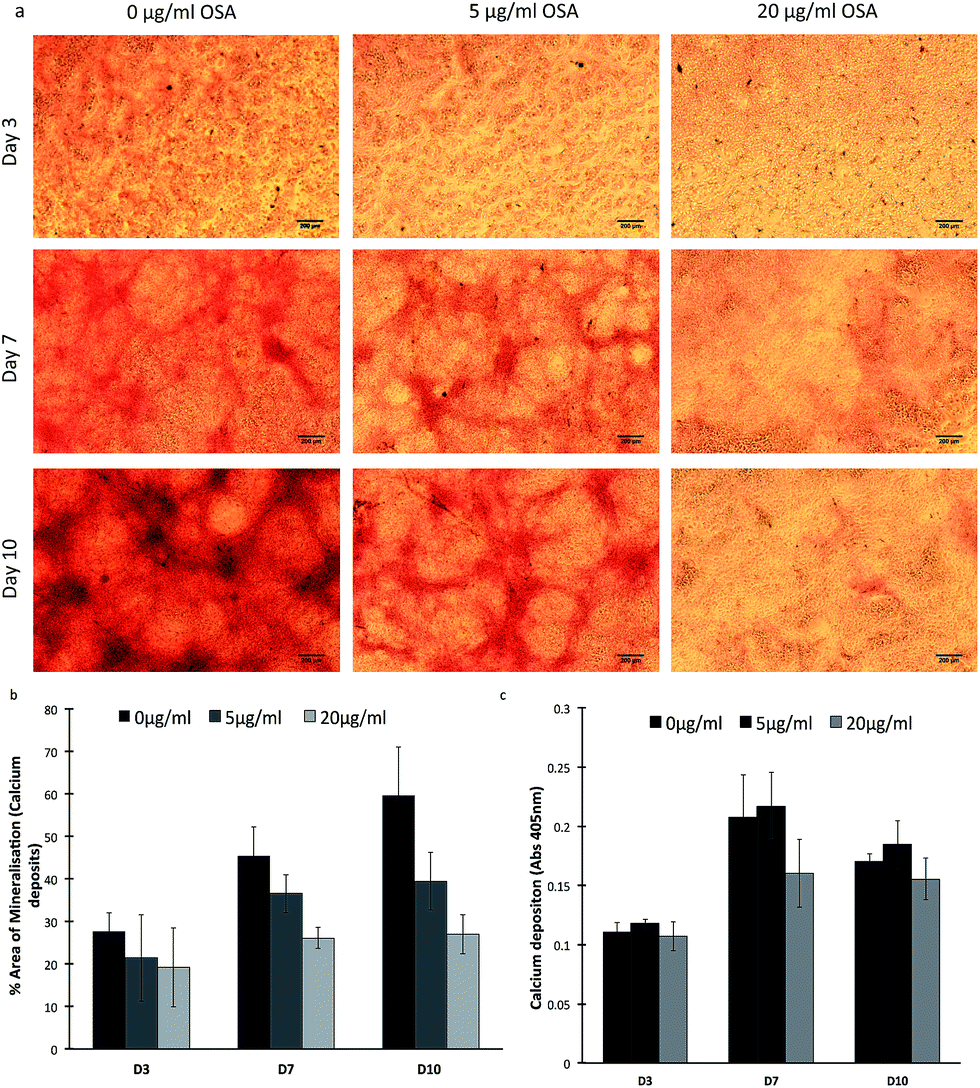 | ||
| Fig. 1 Bone mineral formation was determined using Alizarin Red-S staining in MC-3T3-E1 cultured in the presence of 0, 5 and 20 μg ml−1 OSA. An increase in staining was noted in cultures with 5 μg ml−1 OSA when compared with cultures containing 20 μg ml−1 OSA. Scale bar denotes 200 μm. Semi quantitative analysis (a) indicated that mineral formation was decreased in the presence of OSA. Absorbance measurements (b) after dissolving the stain indicated that there was little difference in total mineral deposition in the different samples when normalized against DNA content. | ||
Thus suggesting that the OSA itself may be involved in mediating the deposition of mineral across the collagen fibrils, giving the impression of a more evenly distributed mineralization of the collagen matrix. Von Kossa staining, used to determine phosphate deposition, also confirmed that the presence of mineral was highest in 5 μg ml−1 OSA treated samples when compared to untreated samples (Fig. S2†). A significant increase (p < 0.05) in mineral deposition was seen in the presence of 5 μg ml−1 OSA when compared to the control. The highest concentration of OSA in the samples showed to inhibit mineral deposition (Fig. S3†). To confirm this observation, samples were ashed at 600 °C and weighed to determine the mass of non-combustible mineral that was present in each culture. Little difference in mineral mass was observed between the samples, suggesting that mineral was present in all OSA containing samples as well as the control (Fig. 2). The presence of mineral in the cultures indicated that the mineral deposition within the cell cultures treated with OSA was of greater homogeneity than in the case of the non-treated specimens, where mineralization was concentrated in fewer discrete sites (Fig. 1c)
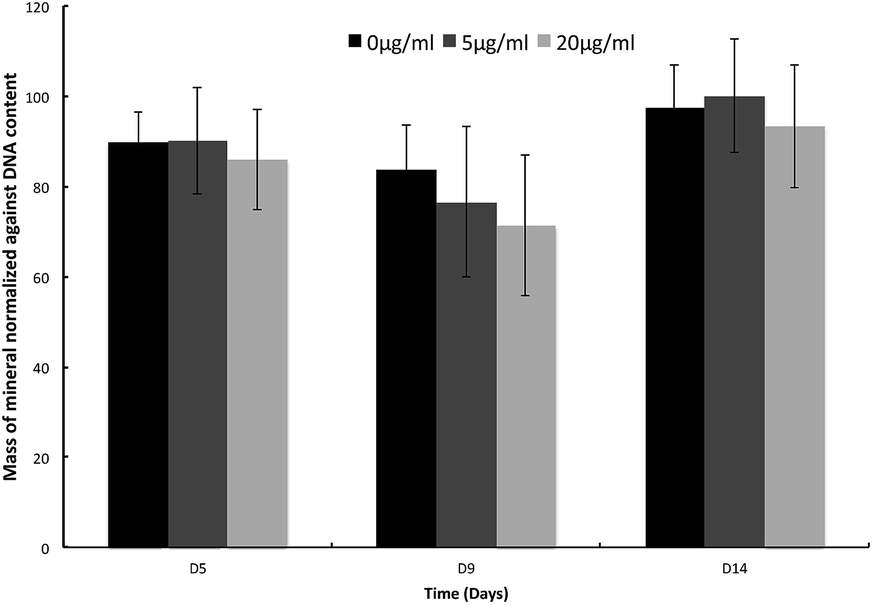 | ||
| Fig. 2 The amount of mineral present in samples with 0, 5 and 20 μg ml−1 OSA was normalized against total DNA content. It was noted that mineral was present in all OSA containing samples when compared to the control, with no significant difference between the samples. | ||
Transmission electron microscopy (TEM) analysis was carried out on collagen gels formed in the presence and absence of OSA medium for 24 h, the gels were added to TEM girds, washed thoroughly with deionised water and left to air dry. The collagen fibrils that had been left in medium for 24 h demonstrated no banding that is typical of mineral deposition (Fig. 3a) and the only sign of mineral was at the surface of individual fibrils and took the form of large and irregular deposits. In stark contrast, the addition of 5 μg ml−1 of OSA (Fig. 3b) to the collagen resulted in the production of intensely and regularly banded collagen fibrils with a periodicity of approximately 70 nm, typical of mineralized bone matrix. The composition of the banded material was determined using selected area electron diffraction (SAED) (Fig. 3d), which suggested that the deposited mineral was apatitic in nature (Fig. 3e–f). This banding arrangement was identical to naturally mineralized type 1 collagen and often attributed to the role of matrix phosphoproteins, which act as templates for the growth of apatite nucleation and growth within the gap zones of collagen fibrils.23 The addition of OSA appeared to replicate the priming of the collagenous matrix for the deposition of the localized mineral.
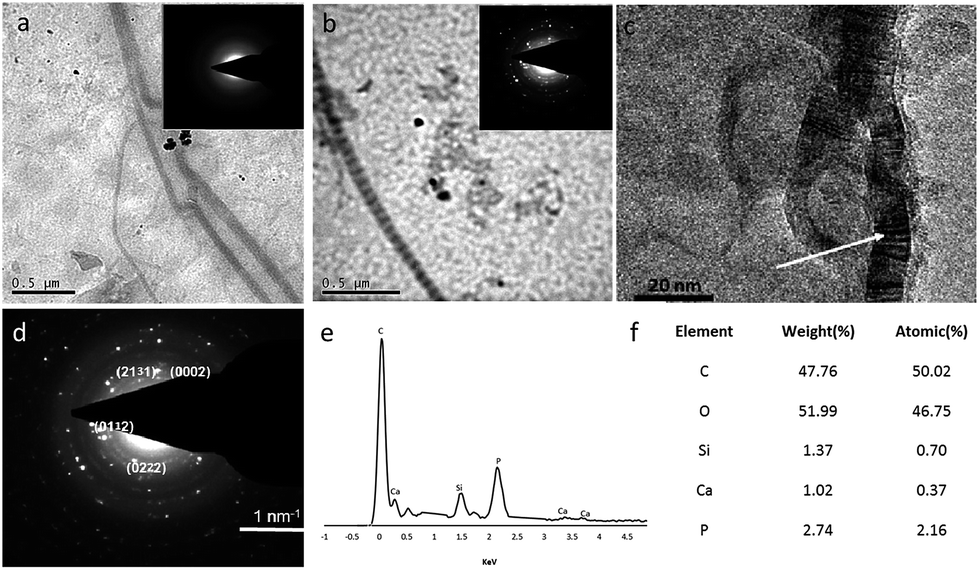 | ||
| Fig. 3 Unstained TEM photomicrographs of collagen gels formed with and without OSA. Collagen fibrils produced without OSA showed irregular mineral deposition on the surface of the collagen fibrils (a). Inset: selected area electron diffraction (SAED) corresponding with those of poorly-crystalline collagen fibrils containing predominantly amorphous electron-dense minerals. The presence of OSA resulted in organised mineral deposition whereby banding was observed (b) and the SAED demonstrated a crystalline phase within the collagen fibrils (inset). HR-TEM of collagen fibrils in the presence of OSA clearly exhibit electron-dense cross-banding within the collagen fibril (arrowed) (c). The composition of the mineral deposition within the fibrils was determined using SAED and energy dispersive X-ray spectroscopy (EDX) and the mineral deposited was apatitic in nature (d–f). | ||
It has previously been hypothesized that a soluble OSA precursor infiltrates the collagen fibrils and condense along the collagen triple helix creating multiple negative silonal groups within the fibrils. This anionic environment within the fibrils sequesters amorphous CaP from the surrounding environment, which is small enough to infiltrate the silicified collagen. In this way the CaP phases are deposited over the original amorphous silica phase.24 Sodium tripolyphosphate (TPP) a linear tripolyphosphate, that has a similar structure to that of OSA, was demonstrated to induce hierarchal mineralization within self-assembling collagen fibrils. TPP forms ionic cross-links (ionic bridges) within collagen molecules leading to the deposition of mineral within the fibrils.22 Thus it can also be proposed that OSA may form ionic cross-links with the collagen as a reversible binding mechanism with the remaining –OH groups contributing to the attraction of amorphous CaP nanocrystals.
Given the striking differences observed between the specimens, the influence of the OSA on the collagen fibril conformation and final diameter was determined using Atomic Force Microscopy (AFM), Circular Dichroism (CD) and turbidity measurements. The addition of OSA to forming collagen gels had a significant influence on the fibril diameter of the collagen shown in Fig. 4. The addition of quantities up to 50 μg ml−1 OSA resulted in a monotonic reduction in fibril diameter and a higher level of fragmentation of the overall structure (Fig. 4b). An increase in diameter when collagen gels were formed with 5–50 μg ml−1 OSA could be attributed to expansion of the lateral spaces between collagen molecules to accommodate the infiltration of the mineral phase.22
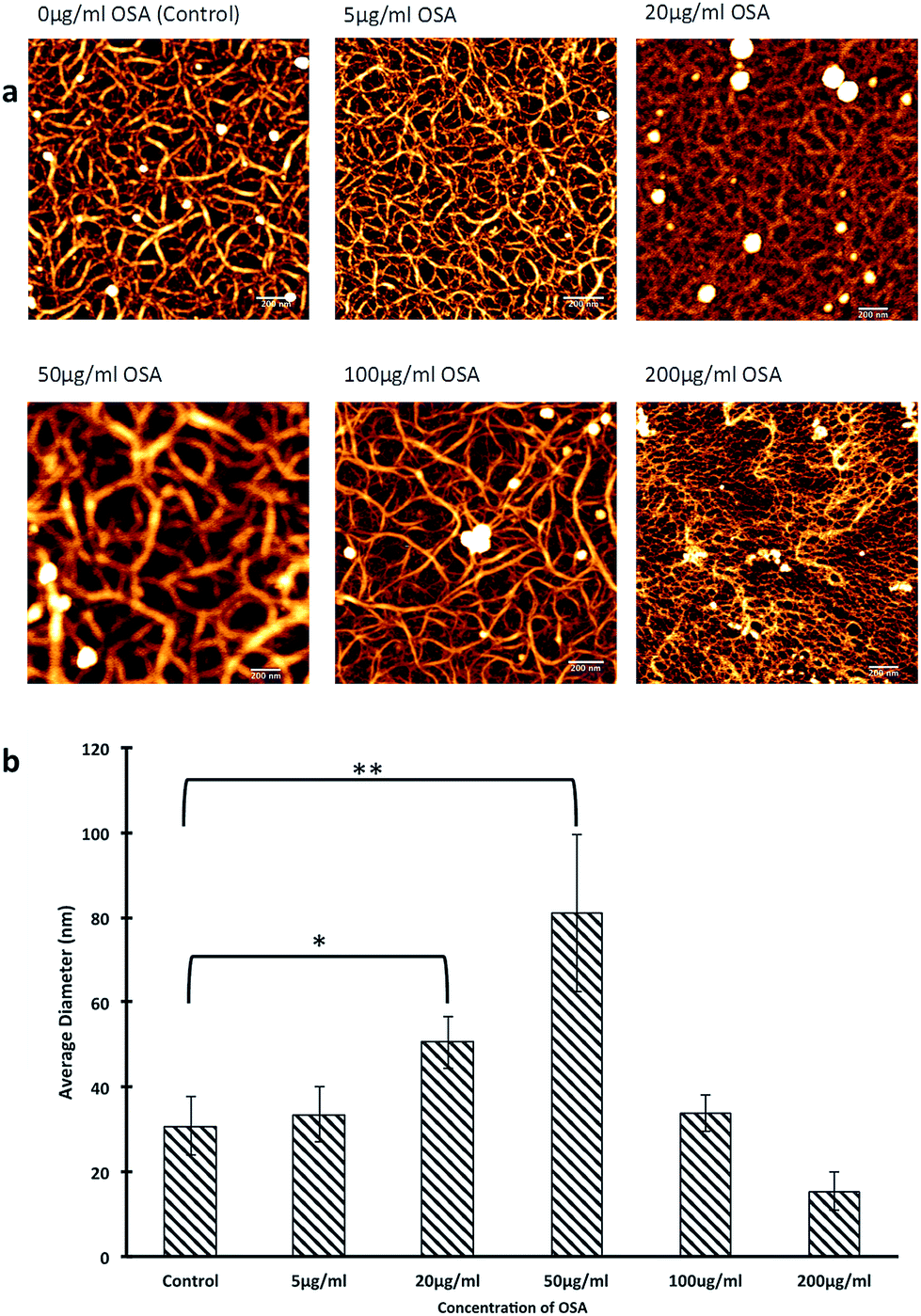 | ||
| Fig. 4 AFM images of collagen fibrils in the presence of various concentrations of OSA. An increase in OSA concentration altered the collagen network and in supra-physiological conditions (200 μg ml−1) OSA completely fragmented the network. Scale bar denotes 200 nm (a) 20–50 μg ml−1 OSA increase fibril diameter although higher levels of OSA however reduced the fibril diameter (b). | ||
It has been suggested that OSA, dissolved from silicon-based glasses, interacts through hydrogen bonding between the specific sites on the protein helices. On the other hand, concentrated OSA, is mainly constituted of negatively charged poly-silicic acids and these interfere with the assembly of triple helices via electrostatic interactions.25 The present study identified a change in morphology of collagen fibrils in the presence of OSA with thicker collagen fibrils forming a more intricate network when OSA was present at concentrations up to 50 μg ml−1. It has been suggested that OSA may induce a conformation change in collagen possibly by modifying the hydration state through changes in hydrogen bonding between helices and localized water molecules; this slows the process down and allows for better packing of the triple helices within the fibrils, generating a compact fibril network.26 It is likely that this mechanism fully explains how the interactions between the collagen molecules and the OSA resulted in a significant change in the morphology of the formed fibrils.27 As the concentration of the OSA was increased, fragmentation of the network occurred, and this was also strong evidence of interactions occurring between the OSA and the collagen fibrils. The 13C and 29Si solid state MAS NMR data from the specimens that were treated with OSA demonstrated the nature of this interaction (Fig. 5). A characterization of the collagen structures using 13C MAS NMR (Fig. 5a) shows that with increasing OSA concentration, there was a monotonic reduction in the 13C resonances ascribed to the carboxylic acid functionalities at ∼δ 170 ppm. These formally quantitative data show that polycondensation is largely complete with the Si speciation in the 100 μg ml−1 OSA sample being dominated by Q4 linkages; this concentration of OSA was the only sample sufficiently concentrated enough to afford a signal-to-noise level that was able to be reliably deconvoluted. This indicated that the OSA molecule interacted with the terminal moieties of the collagen fibril, thus suggesting the creation of a bonding arrangement, which promoted ordered mineralization. In contrast, the more qualitative 29Si CPMAS NMR (Fig. 6b) exhibits an inferior (rather than enhanced) signal-to-noise level despite the polarization transfer from the more abundant/higher γ1H species. This occurrence suggested that highly mobile water molecules were solvating the condensed mineral phase and the 1H–29Si dipolar interaction was modulated and reduced. These finding are of significant importance since it finally elucidates why ionic Si is essential for patterned mineralisation, and it also demonstrates how simple inorganic molecules can have a major influence on extracellular matrix structure which subsequently regulates tissue function.
 | ||
| Fig. 5 13C NMR spectra on OSA/collagen samples indicative of a monotonic reduction in the 13C resonances ascribed to the carboxylic acid functionalities at ∼δ 170 ppm showed that the OSA molecule is interacting with the terminal moieties of the collagen fibril (a) and 29Si NMR spectra on the same samples demonstrated condensed mineral phase as the 1H–29Si dipolar interaction becomes modulated and reduced (b). | ||
 | ||
| Fig. 6 Turbidity measurements of collagen self-assembly with various concentrations of OSA (a). OSA demonstrated a strong influence on the kinetics of collagen fibril formation. CD spectra of collagen formed with different concentration of OSA showed an increase in intensity when collagen gels were formed with 5 and 20 μg ml−1 indicative of an increase in the triple helicity of the structure (b). | ||
To further elucidate how the OSA influenced collagen fibril assembly, turbidity measurements were made and the confirmations of the fibrils were evaluated using circular dichroism. The kinetics of type 1 collagen fibril formation was determined by turbidity measurement as shown in Fig. 6a. The turbidity versus time graphs generated sigmoidal curves with a lag phase before the onset of an increase in turbidity which is indicative of the formation of nano/micro fibrils, followed by a plateau phase resulting in the complete formation of the fibrils. Collagen gels formed with 20 and 50 μg ml−1 OSA had increased lag phases when compared to the collagen gels formed with higher concentrations of OSA (Fig. S4†). Changes in the lag phase indicate that the effect of OSA on collagen fibril formation is most likely due to a modification of the collagen microfibrils before the onset of the self-assembly process. The changes in absorbance are attributed to the fibril thickness and ultimate turbidity of the collagen. No significant difference was observed in turbidity in the presence of 20 μg ml−1 OSA when compared to collagen samples without OSA. A decrease in absorbance in the presence of 50 μg ml−1 OSA is indicative of the formation of thicker fibrils and an increased absorbance in the presence of 100 μg ml−1 OSA demonstrates thinner fibrils as seen in the AFM images in Fig. 5.
The triple helical conformation of native collagen shows a CD spectrum characteristic by a positive band at 223 nm and a negative band at 203 nm, this is in agreement with earlier reported spectra.28 Increases in positive intensities were observed in the presence of 5 and 20 μg ml−1 OSA (Fig. 6b). It has previously been suggested that an increase in the number of hydroxyl groups results in increased stabilization of the protein structure due to the aggregation of collagen molecules.29
The ratio of the positive peak intensity over negative peak intensity (Rpn) was measured to determine the helicity of collagen in the solution, shown in Table 1. An increase in Rpn values was noted as OSA concentration increased, which can be caused by conformational changes in the collagen molecule without complete denaturing of the protein.30
| Concentration of OSA | Maximum ellipticity | Minimum ellipticity | R pn values |
|---|---|---|---|
| 0 μg ml−1 OSA | 1.44 | −7.57 | 0.19 |
| 5 μg ml−1 OSA | 2.42 | −7.10 | 0.34 |
| 20 μg ml−1 OSA | 1.33 | −8.24 | 0.40 |
| 100 μg ml−1 OSA | 2.02 | −3.81 | 0.53 |
Experimental
Alizarin Red-S staining
Alizarin Red-S staining was used to quantify calcium deposits in cultures. 40 mM Alizarin Red-S (Sigma Aldrich, UK) was prepared and adjusted to pH 4.2 with 10% (v/v) ammonium hydroxide (Fischer Scientific, UK). The cultures were fixed with 10% (v/v) paraformaldehyde for 30 minutes at room temperature and washed with PBS. Alizarin Red-S stain was added to the cultures and left at room temperature for 20 minutes. Excess stain was removed and the cultures were washed with PBS. For quantification of the Alizarin Red-S stain, 800 μl of 10% (v/v) acetic acid was added to each well and left to stand for 30 minutes. The cells detached from the bottom of the tissue culture plate and were scraped using a cell scraper (Corning Incorporated, USA) and transferred to a 1.5 ml Eppendorf® tube. After vortexing for 30 seconds the samples were heated at 85 °C for 10 minutes and placed into crushed ice for 5 minutes. The cell slurry was centrifuged at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 minutes and the supernatant removed and transferred to a clean centrifuge tube. 45 μl of 0.1% (v/v) ammonium hydroxide was added to neutralize the acid. An aliquot of the supernatant was transferred to a 96 well plate and read at 405 nm using a plate reader (Promega, UK).31
000 rpm for 15 minutes and the supernatant removed and transferred to a clean centrifuge tube. 45 μl of 0.1% (v/v) ammonium hydroxide was added to neutralize the acid. An aliquot of the supernatant was transferred to a 96 well plate and read at 405 nm using a plate reader (Promega, UK).31
Mass of mineral in cell cultures
MC-3T3-E1 cells were cultured in 6 well plates at a seeding density of 3 × 105 cells per ml in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% (v/v) foetal bovine serum (FBS), 2.5% (v/v) 1 M HEPES, 1% (v/v) 10 mg ml−1 penicillin streptomycin and 2.5% (v/v) 200 mM L-glutamine and left to attach overnight. The following day, supplemented DMEM containing 5 μg ml−1 or 20 μg ml−1 of sodium metasilicate was added to the cells. Total mineral mass was determined over time, at each time point; cells were washed and scraped from the bottom of the wells using a cell scraper. The cell slurry was collected and heated to 600 °C in an oven. The remaining mineral was weighed and normalised against DNA content.Extraction of collagen from rat-tails
Eight- to 10 week-old, male, 175 to 200 g, Wistar rats (Charles River, Kent, UK), housed with free access to food and water, under a 12 hour dark/light cycle were used for rat tail collagen type 1 extraction. Tissue harvest was performed at the Biomedical Services Unit at the University of Birmingham (Birmingham, UK) in accordance with the Home Office guidelines set out in the 1986 Animal Act (UK) and the University of Birmingham ethical review committee and conformed to the Federation of European Laboratory Animal Associations (FELASA) guidelines. Collagen was extracted from rat-tail tendons by removing the outer skin from the tail and pulling out the tendons using sterile forceps. The tendons were solubilized in 0.5 M acetic acid solution for 48 hours at 4 °C. The solubilized products were sterile filtered and stored at 4 °C. Dialysis tubes were prepared by immersing in ethanol (50% v/v) for 1 hour at 40 °C in a fume hood, followed by immersing in 10 mM sodium bicarbonate (NaHCO3)/1 mM EDTA solution at 40 °C for 1 hour. The dialysis tubes were washed in distilled water for 1 hour and stored at 4 °C until required. The solubilized rat-tail extracts were poured into the dialysis tubes and dialyzed against DMEM for 24 h at 4 °C. After dialysis the collagen extract was centrifuged at 10000 rpm for 2 h at 6 °C (Beckman Coulter, UK) and the supernatant was transferred to sterile bottles and stored at 4 °C.
Transmission electron microscopy and evaluating mineral deposition in collagen gels
Transmission electron microscopy (TEM) was performed on collagen gels produced without OSA and with 5 μg ml−1 OSA. 1 μl of the gel solution was added to the carbon grids (Agar Scientific, UK) and left to dry for 30 min. the gels were examined using a Jeol 1200EX TEM (Japan) at 80 kV. All samples were examined without further staining. In order to evaluate the deposition of mineral within collagen fibrils in the presence of OSA and an acellular environment, collagen gels were produced with or without OSA and incubated at 37 °C. The presence of mineralization was determined using, high resolution transmission electron microscopy (HR-TEM), selected area electron diffraction (SAED) and energy dispersive X-ray spectroscopy (EDX) and the peaks corrected for Na and Cl. The obtained SAED patterns were analyzed using Digital Micrograph image (Gatan v3.3) processing software and a computer program for the simulation of polycrystalline electron diffraction patterns and phase identification (JECP/PCED). For the TEM imaging corresponding to the SAED, the samples were analyzed using a FEI Technai 20 TEM (LaB6) at an acceleration voltage of 200 kV in diffraction mode.Atomic force microscopy analysis
Collagen fibril morphology was analyzed using atomic force microscopy (AFM) of gels prepared with different concentrations of OSA and swabbed onto clean silicon substrates before gelling of the samples had occurred. The sample was left to air dry for 20 minutes and rinsed three times in distilled water to remove any excess salts. Surface morphology was determined using a NanoWizard II atomic force microscope (JPK Instruments, UK) operating in intermittent contact mode at a tip velocity of 2 μm s−1, employing pyramidal tipped Si cantilevers (PPP-NCL, Windsor Scientific, UK). Collagen fibril diameter was determined using a NanoWizard II AFM imaging software (JPK Instruments, UK).Nuclear magnetic resonance (NMR)
Both 13C and 29Si MAS NMR measurements were performed at 7.0 T using a Varian/Chemagnetics InfinityPlus spectrometer operating at Larmor frequencies of 75.5 and 59.6 MHz, respectively. All experiments were performed using a Bruker 4 mm HX probe which enabled a MAS frequency of 12 kHz to be enabled. The 13C CPMAS measurements implemented an initial 1H π/2 excitation pulse of 2.5 μs duration, a Hartmann–Hahn contact period of 2 ms and a recycle delay of 2 s. The CP contact was ramped cross-polarisation condition which ranged from 75% to 100% of the 1H channel power, and the 1H decoupling nutation frequency during acquisition was 100 kHz. All 13C chemical shifts were reported against the IUPAC recommended primary reference of Me4Si (1% in CDCl3, δ 0.0 ppm), via an alanine secondary in which the known shift of the CH3 resonance is δ 20.5 ppm. For 29Si single pulse MAS NMR, flip angle calibration was performed on kaolinite from which a π/2 pulse time of 5 μs was measured. All measurements were undertaken with a π/2 tip angle, a recycle delay of 240 s and 1H decoupling during acquisition possessing a nutation frequency of 80 kHz. The 29Si chemical shifts were reported against the IUPAC recommended primary reference of Me4Si (1% in CDCl3, δ 0.0 ppm), via a kaolinite secondary in which the resonance has a known shift of −92.0 ppm.32 The corresponding 29Si CPMAS measurements implemented an initial 1H π/2 excitation pulse of 4 μs duration, a Hartmann–Hahn contact period of 5 ms and a recycle delay of 5 s. This CP contact was also a ramped cross-polarisation condition which ranged from 75% to 100% of the 1H channel power, and the 1H decoupling nutation frequency during acquisition was 80 kHz.Collagen fibrillogenesis
Kinetic analysis of collagen fibril formation was examined in the presence of different concentrations of sodium metasilicate, by measuring the turbidity of the gelling collagen (loss of transparency) over a period of time using spectrophotometry (Cecil Intruments, UK). Collagen gels (3 mg ml−1) were prepared with various concentrations of sodium metasilicate (20 μg ml−1, 50 μg ml−1, 100 μg ml−1) and the pH was adjusted to 8.0–8.5 with 1 M NaOH to promote gelation. 1 ml of the solution was added into a cuvette (1 cm) before gelling of the samples occurred and turbidity-time profiles were determined at an absorbance of 553 nm at 37 °C.Circular dichroism spectroscopy
Circular Dichroism (CD) spectra were recorded in a 0.5 mm path length quartz cuvette at 298 K on a Jasco J-715 spectropolarimeter. Spectra of 0.25 mg ml−1 collagen gel solutions without OSA and different concentrations of OSA were determined. The observed ellipticities in millidegrees were plotted against wavelength (nm). Spectra are an average of 5 scans recorded between 185 nm and 400 nm at 200 nm min−1 with a continuous feeding of the measuring compartment with high purity nitrogen (99.99%) to suppress oxygen absorption.Statistical analysis
Data was subjected to statistical analysis and were reported as mean ± standard deviations. Student paired T-tests between groups and one way analysis of variance (ANOVA) and Tukey post hoc test were used to determine significant differences between groups with p < 0.05.Conclusions
We have for the first time demonstrated that silicon in the form of orthosilicic acid enhances the role of collagen as a template for the intrafibrillar deposition of apatite, something that is likely critical to the mechanical performance of bone and other mineralized tissues. The OSA was shown to interact with the terminal groups in type I collagen molecules, within the hole zones, thus providing a site for ordered mineral deposition to occur. Given the importance of ordered mineralization to the long-term mechanical stability of bone, we believe that this work finally provides a rational explanation for why deprivation of silicon can cause skeletal deformity and its delivery in materials may enhance localized bone growth. Importantly, this work also highlights how even low concentrations of inorganic molecules can have a profound influence on the structural properties of tissues through modification at the molecular and nanoscopic scales.Acknowledgements
J. V. Hanna thanks the University of Warwick, EPSRC and the Birmingham Science City for contributions to the solid state MAS NMR instrumentation used in this research. The latter contribution was partially supported through the Birmingham Science City Advanced Materials Project 1: Creating and Characterising Next Generation Materials Project, with support from Advantage West Midlands (AWM) and partial funding from the European Regional Development Fund (ERDF).References
- E. M. Carlisle, J. Nutr., 1976, 106, 478 CAS.
- K. Schwarz and D. B. Milne, Nature, 1972, 239, 333 CrossRef CAS PubMed.
- R. Jugdaohsingh, S. H. C. Anderson, K. L. Tucker, H. Elliott, D. P. Kiel, R. P. H. Thompson and J. J. Powell, Am. J. Clin. Nutr., 2002, 75, 887 CAS.
- K. Schwarz, Proc. Natl. Acad. Sci. U. S. A., 1973, 70, 1608 CrossRef CAS.
- D. Reffitt, N. Ogston, R. Jugdaohsingh, H. F. Cheung, B. A. Evans, R. P. Thompson, J. Powell and G. Hampson, Bone, 2003, 32, 127 CrossRef CAS PubMed.
- M. Q. Arumugam, D. C. Ireland, R. A. Brooks, N. Rushton and W. Bonefield, Key Eng. Mater., 2003, 32, 869 Search PubMed.
- I. D. Xynos, M. V. J. Hukkanen, J. J. Batten, L. D. Buttery, L. L. Hench and J. M. Polak, Calcif. Tissue Int., 2000, 67, 321 CrossRef CAS PubMed.
- S. Ni, K. Lin, J. Chang and L. Chou, J. Biomed. Mater. Res., Part A, 2008, 85, 72 CrossRef PubMed.
- S. Heinemann, T. Coradin, H. Worch, H. P. Wiesmann and T. Hanke, Compos. Sci. Technol., 2011, 71, 1873 CrossRef CAS.
- N. Patel, S. M. Best, W. Bonfield, I. R. Gibson, K. A. Hing, E. Damien and P. A. Revell, J. Mater. Sci.: Mater. Med., 2002, 13, 321 CrossRef.
- Y. Liu, S. Thomopoulos, C. Chen, V. Birman, M. J. Buehler and G. M. Genin, J. R. Soc., Interface, 2014, 11, 835 Search PubMed.
- A. Biswas, I. S. Bayer, H. Zhao, T. Wang, F. Watambe and A. S. Biris, Bionanocomposites, 2010, 11, 2545 CAS.
- I. S. Bayer, A. Ghosh, M. Labriola and A. B. Biris, RSC Adv., 2013, 3, 20315 RSC.
- Y. Wang, T. Azaïs, M. Robin, A. Vallée, C. Catania, P. Legriel, G. Pehau-Arnaudet, F. Babonneau, M. M. Giraud-Guille and N. Nassif, Nat. Mater., 2012, 11, 724 CrossRef CAS PubMed.
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2010, 78, 929 CrossRef PubMed.
- L. Chen, R. Jacquet, E. Lowder and W. J. Landis, Bone, 2015, 71, 7 CrossRef CAS PubMed.
- D. E. Rodriguez, T. Thula-Mata, E. J. Toro, Y.-W. Yeh, C. Holt, L. S. Holliday and L. B. Gower, Acta Biomater., 2014, 10, 494 CrossRef CAS PubMed.
- J. Sodek, B. Ganss and M. D. McKee, Crit. Rev. Oral Biol. Med., 2000, 11, 279 CAS.
- A. J. Lausch and E. D. Sone, Biomacromolecules, 2015, 16, 1938 CrossRef CAS PubMed.
- M. J. Olszta, E. P. Douglas and L. B. Gower, Calcif. Tissue Int., 2003, 72(5), 583–591 CrossRef CAS PubMed.
- B. Marelli, C. E. Ghezzi, Y. L. Zhang, I. Rouiller, J. E. Barralet and S. N. Nazhat, Biomaterials, 2015, 37, 252 CrossRef CAS PubMed.
- Y. Liu, Y.-K. Kim, L. Dai, N. Li, S. O. Khan, D. H. Pashley and F. R. Tay, Biomaterials, 2011, 32, 1291 CrossRef CAS PubMed.
- A. George and A. Veis, Chem. Rev., 2008, 108, 4670 CrossRef CAS PubMed.
- L. Niu, K. Jiao, H. Ryou, C. K. Y. Yiu, J. Chen, L. Breschi, D. D. Arola, D. H. Pashley and F. R. Tay, Angew. Chem., 2013, 125, 5874 CrossRef.
- D. Eglin, S. Maalheem, J. Livage and T. Coradin, J. Mater. Sci.: Mater. Med., 2006, 17, 161 CrossRef CAS PubMed.
- D. Eglin, K. L. Shafran, J. Livage, T. Coradin and C. C. Perry, J. Mater. Chem., 2006, 16, 4220 RSC.
- S. Heinemann, C. Heinemann, R. Bernhardt, A. Reinstorf, B. Nies, M. Meyer, H. Worch and T. Hanke, Acta Biomater., 2009, 5, 1979 CrossRef CAS PubMed.
- L. Brazdaru, M. Micutz, T. Staicu, M. Albu, D. Sulea and M. Leca, C. R. Chim., 2015, 18, 160–169 CrossRef CAS.
- B. Madhan, V. Subramanian, J. R. Rao, B. U. Nair and T. Ramasami, Int. J. Biol. Macromol., 2005, 37, 47 CrossRef CAS PubMed.
- R. Usha, R. Maheshwari, A. Dhathathreyan and T. Ramasami, Colloids Surf., B, 2006, 48, 101 CrossRef CAS PubMed.
- C. A. Gregory, W. G. Gunn, A. Peister and D. J. Prockop, Anal. Biochem., 2004, 329, 77 CrossRef CAS PubMed.
- R. K. Harris, E. D. Becker and S. M. C. D. E. Menezes, Pure Appl. Chem., 2001, 73, 1795 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra19784a |
| This journal is © The Royal Society of Chemistry 2016 |