
Facile, ethylene glycol-promoted microwave-assisted solvothermal synthesis of high-performance LiCoPO4 as a high-voltage cathode material for lithium-ion batteries†
Abstract
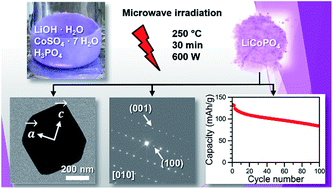
Olivine-type LiCoPO4 is considered a promising high-voltage cathode material for next-generation lithium-ion batteries. However, preparing high-performance LiCoPO4 by a simple approach has been challenging. Herein, we present a facile and rapid (30 min) one-step microwave-assisted solvothermal synthesis route using a 1 : 1 (v/v) water/ethylene glycol (EG) binary solvent mixture and a temperature of 250 °C. The technique delivers high-performance LiCoPO4 nanoparticles without additional post-annealing or carbon coating steps. The as-prepared powder consists of single crystalline LiCoPO4 and features a hexagonal platelet-like morphology with dimensions of 700–800 nm × 400–600 nm × 100–220 nm. Selected area electron diffraction (SAED) experiments reveal that the platelets show the smallest dimension along [010], which is the direction of the lithium diffusion pathways in the olivine crystal structure. Furthermore, the results indicate that the EG co-solvent plays an important role in tailoring the particle size, morphology, and crystal orientation of the material. Co L-edge soft X-ray absorption spectroscopy (XAS) of LiCoPO4 are presented for the first time and confirm that the material only consists of Co2+. Benefiting from the unique morphology, which facilitates Li-ion conduction, electrochemical measurements deliver an initial discharge capacity of 137 mA h g−1 at 0.1 C, a remarkably stable capacity retention of 68% after 100 cycles at 0.5 C, and a specific energy density of 658 W h kg−1 based on its capacity and voltage, which is the best performance of LiCoPO4 obtained from microwave-assisted solvothermal synthesis to date.
 Please wait while we load your content...
Please wait while we load your content...

