
Multiple functionalization of tungsten disulfide inorganic nanotubes by covalently grafted conductive polythiophenes†
Abstract
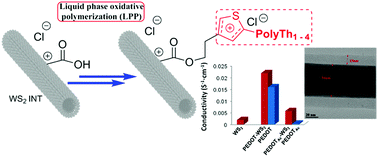
According to the “polymer growth from surface” concept, a variety of dual phase polythiophene–tungsten disulfide inorganic nanotube (PTh–WS2 INT) composites have been successfully prepared. This concept has made use of a covalent approach of liquid phase oxidative polymerization (LPP) directly from a WS2 INTs surface. In contrast to a simple bulk LPP, this oxidative polymerization variant enables an effective way to control the interfacial polythiophene-based layering. Various chemically stable nanometric polyTh adlayers have been generated towards morphologically well-defined core–shell WS2 INTs/polymer composites. The introduced free carboxylic acid groups, arising from the COOH-functionalized thiophene monomers, seriously improve the functional WS2 INTs dispersability, and thus, provide a versatile route for additional step surface modifications. This study has opened the attractive possibility of achieving high charge mobility via a two-dimensional transport through the WS2 INTs/polymer interface, combined with mobility anisotropy through the crystalline domains of grafted polymer shells.
 Please wait while we load your content...
Please wait while we load your content...

