
Ordered intermetallic Pt–Cu nanoparticles for the catalytic CO oxidation reaction†
Abstract
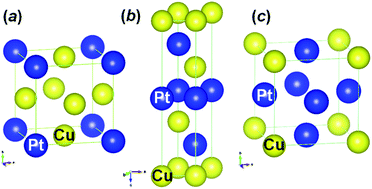
Platinum-based intermetallic nanoparticles (NPs), using the abundantly available element copper, with an average particle size of 4–5 nm on a γ-Al2O3 support were prepared successfully to reduce the consumption of Pt for the removal of CO through the catalytic oxidation reaction from flue gases. Intermetallic Pt–Cu NPs (Pt3Cu, PtCu, and PtCu3) with a Pt loading weight of 5 wt% were prepared on the γ-Al2O3 support by a simple wet impregnation method followed by calcination at various temperatures (500–800 °C) in a H2 environment and they were characterized by powder X-ray diffraction analysis (pXRD), high resolution transmission electron microscopy (HR-TEM), selective area electron diffraction (SAED) method, etc. Despite the higher synthesis temperature of these intermetallic NPs, they were not agglomerated and formed a highly ordered intermetallic structure. The surface of the intermetallic Pt–Cu NPs with cubic-type structure (Pt3Cu and PtCu3) is enclosed of {200} facets, regardless of the significant difference in their compositions. Whereas the surface of rhombohedral-type intermetallic PtCu NPs is enclosed of {104} facets. Although the Pt-loading weight of these intermetallic NPs was the same, Pt3Cu NPs showed a stable and enhanced catalytic activity compared to the other intermetallic PtCu and PtCu3 NPs. Pt3Cu NPs showed an onset and maximum conversion temperature of 50 and 125 °C, respectively. The intermetallic phase between Pt and Cu of Pt3Cu NPs did not decompose; however, the intermetallic phase did decompose for PtCu and PtCu3 NPs after catalytic CO oxidation. Unlike PtCu and PtCu3 NPs, the Pt3Cu NPs were not agglomerated and they were finely dispersed even after catalytic CO oxidation.
 Please wait while we load your content...
Please wait while we load your content...

