
Chiral dendrigraft polymer for asymmetric synthesis of isoquinuclidines†
G. Smitha and
K. Sreekumar*
Department of Applied Chemistry, Cochin University of Science and Technology, Kochi-22, Kerala, India. E-mail: kskpolymer.cusat@gmail.com; Tel: +91-484-2575804 Tel: +91-944-7121530 Tel: +91-944-7053021
First published on 2nd September 2016
Abstract
The synthesis of multi-arm star dendrigraft amidoamine polymer with pentaerythritol initiated polyepichlorohydrin as the core, PEN-G2, on a solid resin support has been demonstrated. We have functionalized a Merrifield resin support with polyepichlorohydrin, and dendrons are grown on it up to second generation using a “graft from” approach. The estimation of amine functionality was done and found to be 25 mmol g−1 which was 70% of the theoretical value. Photolytic cleavage of the dendrigraft polymer from the support resulted in a brown water-soluble viscous liquid. The molecular weight of 3966 shows that out of the 9 G1 dendritic substituents, 5 of them were converted to generation G2. The TEM image of the G2 polymer shows an inhomogeneous polygon-like appearance. The chiral modification of the dendrigraft polymer, PEN-G2, has been done. The copper complex of chiral modified PEN-G2 dendrigraft polymer was synthesized and characterized. The copper complex of PEN-G2 dendrigraft polymer was found to be an excellent catalyst for the synthesis of isoquinuclidines via aza Diels–Alder reaction between cyclohexenone and imines. The catalyst exhibited superior enantioselectivity. The reusability of the catalyst was studied for five reaction cycles.
1. Introduction
The term dendronized or dendrigraft polymer has emerged as a new concept in macromolecular design and materials science due to the unique structures which are seldom achieved by dendrimers and linear polymers.1 The term dendronized polymer is assigned to polymers having a relatively linear core while the term dendrigraft polymer is applicable to branched core polymers. Reactive oligomers or polymers are used to produce dendrigrafts or dendronized polymers, instead of using monomers, as if in constructing dendrimers. The lengthy purification procedure for the synthesis of these dendritic polymers directs us to use the “solid phase strategy”. Solid phase synthesis is really a challenge, because of the large number of reactions that take place simultaneously during the building up of each generation and the number increases exponentially with increase in generation. Conversely, solid phase synthesis provides some inherent advantages over orthodox solution synthesis.2 Synthesis of dendritic polymers need large excess of reagents for obtaining high purity and defect free products and solid phase synthesis usually use large excess of reagents. Purification of intermediates is easy in solid phase synthesis, which is the most difficult and time-consuming process in solution phase synthesis of dendritic polymers. In addition, the solid resins on which the dendritic polymer is synthesized can be used as a highly functionalized heterogeneous catalyst3 and also as highly loaded resin for combinatorial library synthesis, with particular emphasis on one bead one compound approach.4Recently, we reported solid phase synthesis of PAMAM dendrigraft polymer having glycerol initiated polyepichlorohydrin core, which was the first dendrigraft polymer on insoluble support.3 In 1997, Bradley and co-workers introduced the solid phase synthesis of PAMAM dendrimers on organic polymer support.5 Initially, the dendrons were prepared on Tentagel® up to the fourth generation. The assembly of the dendrons followed the divergent synthetic strategy developed by Tomalia et al. for PAMAM dendrimers in solution.6 It was based on alternating steps of double Michael addition of terminal primary amines to methyl acrylate and aminolysis of the terminal esters formed with 1,2-alkanediamines. Later, the approach was extended to PAMAM dendrons on polystyrene beads equipped with a short PEG spacer.7 The reaction proceeded smoothly for the synthesis of first and second generation dendrimers. Third and fourth generation dendrimers showed defect structures as observed from ESI MS data. These dendritic resins were used as high load resins for the synthesis of various molecules.8 The present approach to synthesize dendrigraft polymers includes similar strategy of Michael addition and amination, but follows fewer steps and more number of branches compared to conventional PAMAM dendrimer. To the best of our knowledge, this is one of the few attempts to synthesize dendrigraft polymers having branched polymeric core on a resin support other than the one reported earlier.3 Most of the reports were about single core high generation dendritic systems.9 So far, no report has appeared on the synthesis of amidoamine polymer on a polymeric core. Our aim was to synthesize dendritic systems having large amount of functionality even in the low generation level and we have adopted a ‘graft from strategy’ to synthesize these systems using polymeric core on Merrifield resin.
The amine capacity of Merrifield resin supported PAMAM dendrimers reported earlier, was found to be between 1 and 2 mmol g−1 for G0 to G3 generations.9n So here, we have tried to increase the amine capacity even at the low generation level by incorporating an oligomeric core. For this purpose, we have selected pentaerythritol initiated polyepichlorohydrin (PECH) as core, coupled to the Merrifield resin and amidoamine dendrons were made to grow on it. As a result large number of amidoamine groups can be incorporated in a single step as well as in low generation. After the facile synthesis, photolytic cleavage of the resin gives a broad understanding of the structure of the synthesised polymer.
Asymmetric reactions using chiral catalysts provide the most promising method for construction of chiral molecules.10 Catalytic aza Diels–Alder reaction is one of the useful methods for the synthesis of chiral nitrogen containing heterocycles.11 Using aza Diels–Alder reaction, we can synthesize isoquinuclidines (azabicyclo [2.2.2]octanes) having N-bicyclic structures. They are the structural elements of numerous naturally occurring alkaloids with interesting biological properties.12 A number of chiral Lewis acids and chiral Brønsted acids, as organocatalysts were reported for the activation of imine functional groups.13 In particular, chiral phosphoric acid based organocatalyst was used for the activation of imine functional group which resulted in a number of asymmetric additions of various nucleophiles to imines.14 In 1996, Kobayashi et al. reported the first examples of catalytic asymmetric aza Diels–Alder reaction using a chiral ytterbium catalyst.15 Later, they reported13d,13f,16 that in the presence of 1–5 mol% of Merrifield resin supported chiral BINOL zirconium catalyst, aza Diels–Alder reaction proceeded smoothly to afford the corresponding piperidine derivatives in high yields with high enantioselectivities.13e Silane Lewis acid is effective for the enantioselective promotion of aza Diels–Alder reaction of acylhydrazones with Danishefsky's diene, adding to the versatility of the family of silicon Lewis acids.17 An efficient synthesis of hexahydropyrrolo[3,2-c]quinolin-2-ones and hexahydropyridino[3,2-c]quinolin-2-ones has been developed in moderate to high yields by one-pot two-step aza Diels–Alder reaction. The hexahydropyrrolo[3,2-c]quinolin-2-ones were formed as a single exo-stereoisomer in most cases and hexahydropyridino[3,2-c]quinolin-2-ones were formed as a mixture of exo- and endo-isomers favoring the endo-diastereomer.18 The chiral N,N′-dioxide–Yb(OTf)3 complex-catalyzed enantio-selective aza Diels–Alder reaction of Brassard's diene with aldimines has been developed, giving the corresponding α,β-unsaturated δ-lactam derivatives in moderate yields with good enantioselectivities under mild conditions. Isolation of the reaction intermediate indicates that this asymmetric aza Diels–Alder reaction proceeds through a stepwise Mannich-type pathway.19 Catalytic asymmetric Diels–Alder reaction of α-amino-o-quinodimethane with α,β-unsaturated aldehydes was achieved with high diastereo- and enantioselectivities in the presence of L-proline, which acts as a promoter to generate the quinodimethane from the corresponding precursor as well as a chiral catalyst.20
To date, there have been only a few reports regarding direct aza hetero-Diels–Alder reaction of cyclohexenone with aromatic aldimines. The amino acid catalyzed asymmetric aza Diels–Alder reaction between aqueous formaldehyde, unsaturated cyclic ketones, and aromatic amines furnished the desired azabicyclic ketones (24–48 h) with >99% ee.21 A chiral phosphoric acid, derived from 3,3-di(4-chlorophenyl)-H8-binol, exhibited superior enantioselectivity, affording fairly good yields for the reaction of a range of aromatic aldimines with cyclohexenone within 6 days.22 Co(III) anionic complexes of Schiff bases obtained from salicylaldehydes and enantiomerically pure amino acids act as a novel chiral Bronsted acid to catalyse aza Diels–Alder reaction in CCl4.23
Polymer-supported Lewis acids are known to catalyze cycloadditions and polymer-loaded or dendrimer-loaded copper catalysts have been reported to be promoters of Diels–Alder reactions.24 Menger et al. described the synthesis of polystyrene-based metallo-polymers containing copper and their use as catalysts in Diels Alder reaction.25 Results suggest that metallo-polymers give better yields than the respective metal complex. Chow et al. reported the synthesis of dendritic bis(oxazoline)/Cu(II) complexes and their use as catalysts in the Diels–Alder reaction.26 The size of the dendrimers had a strong influence on the kinetics of the reaction with a possible folding-back of the dendritic sectors towards the metallic site for higher-order dendrimers. Fujita et al. reported the use of dendritic bipyridines Dn as ligands for Cu(OTf)2 in the Diels–Alder reaction of various dienes and dienophiles.27
While, rather fruitful results have been reported in catalytic asymmetric Diels–Alder reactions,10,11,23,28 successful results are limited in asymmetric aza Diels–Alder reactions using dendritic systems. Amino acid derivatives were well-known for enantioselective Diels–Alder reaction.13m,29 These results30 have led us to investigate whether an amino acid–metal complex of dendritic system could be able to mediate the classical aza Diels–Alder reaction through a catalytic enamine mechanism. Herein, we report the direct catalytic aza Diels–Alder reaction of cyclohexenone with aldimines, achieved with good diastereo and enantioselectivities in the presence of a copper complex of proline modified dendrigraft polymer supported on Merrifield resin.
2. Results and discussion
2.1 Characterization of PEN-G2 polymer
The resin supported dendrigraft azide polymer was synthesized using the schematic procedure (Scheme 1). | ||
| Scheme 1 Synthesis of resin supported azide polymer. | ||
The branched hydroxyl terminated PECH (polyepichlorohydrin) was prepared by the ring opening polymerization of the oxirane group in ECH (epichlorohydrin) in the presence of pentaerythritol by activated monomer mechanism (AMM).31 It was a light yellow viscous liquid. From GPC, the molecular weight of PECH was found to be 1037 with polydispersity index of 1.27. The 1H NMR spectrum indicated that the hydroxyl groups at the terminal ends were secondary in nature.
Merrifield resin (1% cross-linked with DVB) was gifted from Thermax India Ltd. Its chlorine capacity was found to be 5.2 mmol g−1. It was made photoactive by the introduction of a nitro group at the ortho position of chloromethyl group.32 The PECH was coupled to the resin using sodium hydride and tetrabutyl ammonium bromide.33 After coupling, chlorine capacity was found to be 7.2 mmol g−1 by Volhards method. From the scanning electron micrographs of nitrated Merrifield resin and PECH attached Merrifield resin, it was found that, after modification with PECH, beads with low extent of grafting were spherical with an appearance similar to the precursor resin particles. SEM EDAX showed that 18 atom% of chlorine was present in the sample.
The IR spectral data of nitrated Merrifield resin at 1596 cm−1 and 1360 cm−1 correspond to the stretching vibrations due to NO2 group. The IR spectrum of PECH coupled Merrifield resin showed a band at 1107 cm−1 and a broad band at 3430 cm−1 which corresponded to C–O–C stretching of ether linkage and due to the hydroxyl group of polyepichlorohydrin. In solid state CP MAS 13C NMR spectrum, broad signal around 50–85 ppm includes CH2–OH, CH–O–CH2 carbons of polyepichlorohydrin, the signal around 125 to 150 ppm is due to carbons of polystyrene support and 0–50 ppm includes carbons due to alkyl part of both support and PECH (Fig. 1).
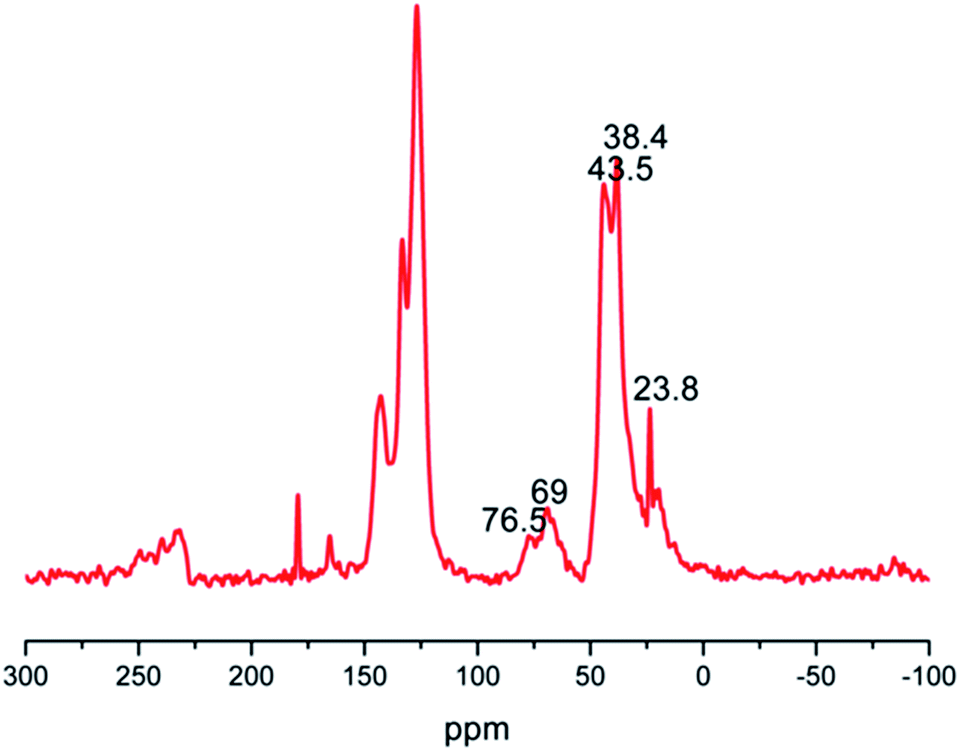 | ||
| Fig. 1 Solid state CP MAS 13C NMR spectrum of PECH attached Merrifield resin. | ||
In the TG curve of PECH attached resin, two mass loss steps were observed. The first decomposition corresponded to nearly 54% mass loss in the temperature around 323 °C. The second stage was completed around 413 °C. The first step of mass loss in TG could be attributed to elimination of chlorinated compounds. The mass loss above 400 °C was due to the breakdown of the polymer backbone (Fig. 2).34
 | ||
| Fig. 2 TG-DTG plot of PECH loaded Merrifield resin. | ||
These observations give clear evidence for the attachment of PECH to the Merrifield resin. The hydroxyl groups of PECH loaded resin was estimated quantitatively. The amount of hydroxyl groups was found to be 1.86 mmol g−1 of the polymer. The hydroxyl group of the polymer was reacted with p-toluene sulphonyl chloride (TsCl) in pyridine.35 After tosylation, percentage of sulphur was found to be 2.44% from CHNS data (C-68.39%, H-5.66%, S-2.44% and N-1.55%). The disappearance of broad deep peak around 3500 cm−1 in the IR spectrum of tosylated PECH indicated the alteration of hydroxyl group to tosyl group. During tosylation, it was observed that the yield was decreased to half by keeping the reaction for more time. This was due to the cleavage of the ether linkages in the polymer by the reaction with TsCl.36 The situation was same when 4% cross-linked Merrifield resin was used. The reaction was also monitored with p-toluene sulphonyl chloride in triethylamine. But there was no great noticeable change with respect to the yield of the product. The azidation of tosylated PECH was carried out using sodium azide in dimethylformamide at 85–90 °C.37 An intense peak at 2100 cm−1 in IR spectrum indicated the presence of azide functionality. In the solid state CP MAS 13C NMR spectrum, due to the conversion of CH2–Cl of PECH to CH2–N3, the signal at 43.5 ppm was shifted to 38.5 ppm. The colour of the resin was changed to brown. The polyazide on reduction with lithium aluminium hydride in THF got converted to –CH2NH2 (Scheme 2).38
 | ||
| Scheme 2 Synthesis of G0.5 dendrigraft polymer. | ||
In the IR spectrum of G0 polymer, the peak at 2100 cm−1 had completely disappeared and a peak at 3451 cm−1 showed the presence of amine functionality (Fig. 3). The quantitative estimation showed the presence of 9.125 mmol of amine per gram of the resin. This high amine capacity compared with previously reported polyamines could happen only if the resin was loaded with PECH.
 | ||
| Fig. 3 IR spectra of PEN-G0, PEN-G0.5, PEN-G1, PEN-G1.5, PEN-G2. | ||
Quantification of the number of amino groups from DTG result was very close to the value obtained by analytical methods (Fig. 4).39 In the TG trace of G0 polymer, the first mass loss of about 20% at 186 °C was due to the elimination of amine as molecular nitrogen. The second mass loss of about 40% at 404 °C was due to the degradation of polyether chains. The Michael addition of the G0 polyamine with methyl acrylate in methanol showed a peak at 1738 cm−1 in the IR spectrum of G0.5 (Fig. 3).6 Even though IR spectrum showed vibration bands in the region around 3400 cm−1, negative Kaiser ninhydrin test result confirmed the absence of free amino groups.
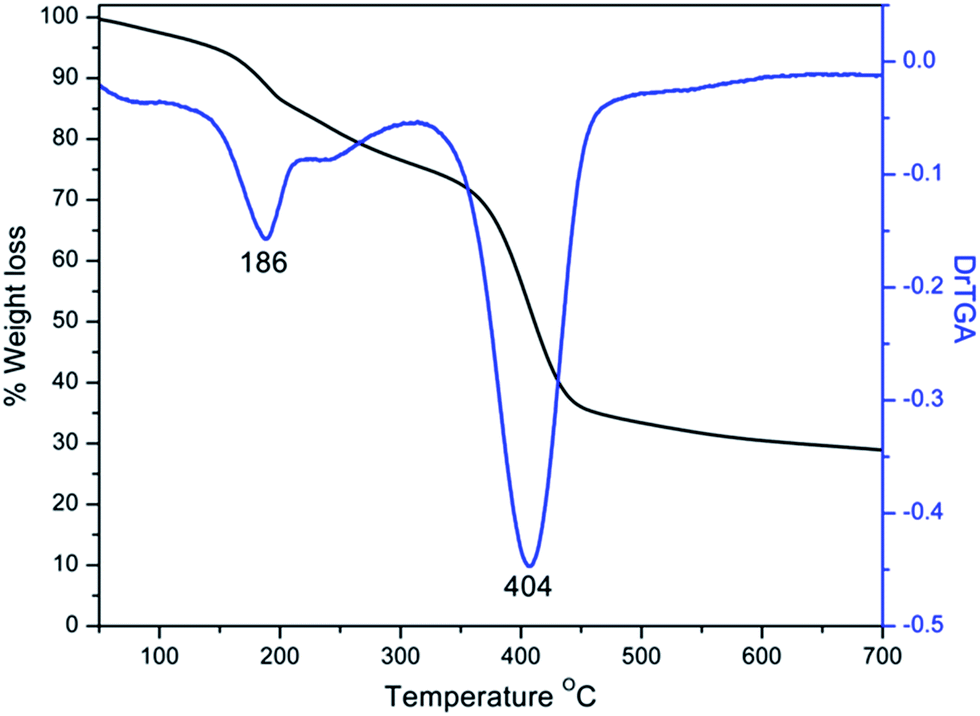 | ||
| Fig. 4 TG-DTG plot of PEN-G0. | ||
In the solid state CP MAS 13C NMR spectrum of G0.5, appearance of a peak at 169 ppm confirmed the incorporation of ester functionality. The G0.5 polymer on amination with ethylene diamine got converted to amidoamine G1 polymer (Scheme 3).40
 | ||
| Scheme 3 Synthesis of G1 and G2.0 dendrigraft polymer. | ||
In the IR spectrum of G1 polymer, bands at 1641 and 3466 cm−1 are due to the amide carbonyl and amine moiety of the polymer (Fig. 3). Both Michael addition and amination reaction took prolonged time for completion because of the poor swelling of resin loaded polymer in methanol. The amine capacity of G1 polymer was found to be 18.75 mmol g−1. In the TG curve of G1 resin; two mass loss steps were observed. The first decomposition corresponded to nearly 26% mass loss in the temperature around 200 °C. The second stage mass loss of 30% was observed around 383 °C (Fig. 5). The first step of mass loss in TG can be attributed to elimination of molecular nitrogen form the amino groups. The mass loss near 400 °C was due to the thermal degradation of the polymer backbone leading to the formation of hydrogen, carbon monoxide, carbon dioxide, methane, ammonia, hydrogen cyanide gas and other higher hydrocarbons.34
 | ||
| Fig. 5 TG-DTG plot of PEN-G1. | ||
The synthesis procedure was repeated to get G2 DP (dendrigraft polymer). Michael addition of G1 polymer resulted in the conversion of G1 to G1.5 dendrigraft polymer. The peak at 1641 cm−1 in the IR spectrum due to carbonyl stretching of amide –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of PEN-G1 got converted to 1740 cm−1 after Michael addition with methyl acrylate (Fig. 3). In the solid state CP MAS 13C NMR spectrum of PEN-G1.5, the signal at 169.4 ppm corresponds to ester carbon (Fig. 6).
O of PEN-G1 got converted to 1740 cm−1 after Michael addition with methyl acrylate (Fig. 3). In the solid state CP MAS 13C NMR spectrum of PEN-G1.5, the signal at 169.4 ppm corresponds to ester carbon (Fig. 6).
 | ||
| Fig. 6 Solid state CP MAS 13C NMR spectrum of PEN G1.5 polymer. | ||
The above G1.5 polymer on amination with ethylene diamine was converted to G2 polymer. The IR spectrum of PEN-G2 polymer showed vibration band at 1662 cm−1 due to carbonyl stretching of amide carbonyl and the band at 1740 cm−1 in the G1.5 due to ester functionality was disappeared (Fig. 3). TG plot of PEN-G2 polymer showed similar decomposition pathway as that of G1 polymer, but a total mass loss of 36% at 194.7 and 272.4 °C, and a 25% mass loss at 407 °C had occurred (Fig. 7).
 | ||
| Fig. 7 TG-DTG plot of PEN-G2. | ||
Scanning electron micrograph of G2 DP shows irregular agglomerates of different shapes (Fig. 8). The loss of spherical nature was due to the crushing of the supported dendrigraft polymer due to prolonged stirring or may be due to the umbrella effect (to stretch to maximum extent) of the resin supported dendritic polymer.
 | ||
| Fig. 8 SEM image of PEN-G2. | ||
The amine capacity of G2 polymer was found to be 25 mmol g−1 which was not in good agreement with the theoretical value. This discrepancy is probably due to defective growth of the dendrons as a result of steric crowding and also due to hydrogen bonding which results in the lack of availability of free amine functionality for carrying out the Michael addition and subsequent reactions and also to a small extent of interpenetration of branches to one another. The amount of amino group in the G0, G1 and G2 serious was found to be higher for GLR-Gn polymer than PEN-Gn polymer.3 This may be due to the monodispersity and high molecular weight of glycerol initiated polyepichlorohydrin compared to pentaerythritol initiated polyepichlorohydrin, the one reported here.
2.2 Characterization of photolytically cleaved PEN-G2 dendrigraft polymer
Under photolytic conditions, the G0, G1 and G2 dendrigraft polymer undergo cleavage from the support. On photolytic cleavage, the nitro group on polystyrene resin was converted to nitroso group with the elimination of dendritic polymer (Scheme 4).41 | ||
| Scheme 4 Mechanism of photolytic cleavage. | ||
The resultant G0 polymer was a brown viscous liquid, soluble in DMSO and methanol. The Chem Bio 3D image of G0 polymer cleaved from the support is shown in the Fig. 9.
 | ||
| Fig. 9 Chem Bio 3D image of G0 dendrigraft polymer. | ||
The 1H NMR spectrum shows that the polymeric arms are somewhat similar. The appearance of peaks in the range 3.2–3.9 ppm in the 1H NMR spectrum is due to the CH2–O, CH–O and NH2 protons of the polymer. The observed behaviour suggests that growth is regulated by a feedback mechanism due to confinement imposed by the support.42 From MALDI TOF MS analysis, the molecular weight of the polymer was found to be 884, which was agreeable with the theoretical value (Fig. 10).
 | ||
| Fig. 10 MALDI MS spectrum of PEN-G0 polymer. | ||
Resin loaded G1 polymer on photolytic cleavage gives a brown viscous liquid. One issue that needs to be addressed is the solubility of dendronized polymers (DP). It has been known that, in general, DPs containing large number of free peripheral amine groups have low solubility in most commonly used solvents.43 The lower solubility of the branched chains was attributed to their high propensity to crystallization.1l Inspection of the 1H NMR spectrum of the G1 DP cleaved from the resin clearly indicates the appearance of amide proton at 8.1 ppm which was absent in G0 DP. To determine the exact number of dendritic substituents introduced, MALDI-TOF MS analysis was performed (matrix: dihydroxybenzoic acid) with the dendronized PEN-G1 polymer. But polymers with polydispersity index greater than 1.2 are difficult to be characterized with MALDI MS due to the signal intensity discrimination against higher mass oligomers.44 The MALDI-TOF MS of PEN-G1 exhibited several peaks. Focus may be given to two major peaks. The peak at 2857.55 Da is derived from nine dendritic substituents on a pentaerythritol initiated polyether chain. The base peak at 1664.39 Da is derived from four dendritic substituents on a pentaerythritol initiated polyether chain. These results indicate that the G0 polymer was modified to ∼70% with dendron substituents on the polyether backbone. Generally, in order to minimize defects or cyclisation and to ensure complete reaction, as we go from lower to higher generation, large molar excess of the reagents have to be taken.45
To confirm the introduction of dendritic substituents onto polyether, dendronized G1 polymer was analyzed by atomic force microscopy (AFM).46 Samples for AFM analysis were prepared by spin coating methanolic solution of the dendronized polymer on a glass plate and AFM analysis was performed in the tapping mode. The AFM image of the film of G1 polymer deposited from higher concentration (0.1% w/w) at a scan width of 75 μm shows spherical aggregates of different sizes.
The 1H NMR spectrum of PEN-G2 dendronized polymer shows broadening of peaks in comparison with PEN-G1 DP. The occurrence of structural defects and dendritic aggregation due to the interdigitation of dendritic arms are commonly encountered during the synthesis of high generation dendritic polymers.47 In this regard, defects in the dendritic structure can arise through incomplete reactions brought about by steric congestion at the periphery or backfolding of flexible dendritic branches, which hides their functional groups. The highest molecular weight of the PEN-G2 polymer from MALDI MS analysis was found to be 3966.02 with a base peak at 1225.41 and another major peak at 1662.72. The molecular weight at 3966.02 shows that out of the 9 G1 dendritic substituents, 5 of them got converted to G2.
The peak at 1662.72 corresponds to the formation of one G2 dendron unit. The peak at 1225.41 is due to cleavage of G2 dendritic unit from the polymeric backbone. The highest mass of 3966 shows that only 30% of dendron substituents are incorporated. The low experimental mass compared to theoretical mass can be due to missing branches. The difference in experimental and theoretical masses can partially be attributed to structural defects, because, errors can also be due to the polydisperse nature of the polymer. Also, the lower mass may be due to the thermal decomposition of the high generation polymer in comparison with lower generation dendrigraft polymers.48 For a comparison we tried to find out the mass of the glycerol based dendrigraft GLR-G2 polymer.49 Merrifield resin carrying the GLR-G2 polymer was cleaved photolytically. The molecular weight obtained from MALDI analysis was 2927, which corresponded to 9 dendritic units on the polyether backbone.
Molecular simulation of the PAMAM dendrimers have indicated that the early full generations (G = 1–2) are characterized by asymmetric disk like shapes. These correspond to open structures that can be readily penetrated by solvent. Higher generations possess a nearly spherical shape, which corresponds to closed, densely packed surface structures.50 In contrast to dendrimers, dendrigraft polymers show more closed or spherical nature at low generation, but at higher generations, polymeric arms are more stretched and have rod like appearance.51
The shape of the polymer in the case of PEN-G2 was analyzed by atomic force microscopy (AFM) as in the case of PEN-G1. In AFM, the film uniformity is mostly determined by the concentration of the dendritic solution regardless of generation.52 The AFM image of the G2 polymer from lower concentration (0.01 wt%) at a scan width of 15 μm around 10 nm shows a small elongated worm or string like appearance. Since polyether backbone is only having approximately 10–13 units, the length of the worm that we observed is agreeable with the length of the polyether oligomer. Despite the approximate nature owing to uncertainties associated with the AFM technique (tip convolution), it was clear that the average worm length corresponds to the length of the parent polyether, whereas the height of the polymer chain depends upon the dendron generation.53
The TEM micrograph of the PEN-G2 polymer shows polygon shape with low molecular weight polymers aggregate inside the polygon (Fig. 11). The polygon shape with longest-extension diameter of up to 13.7 nm may be due to the clustering of polymeric arms in solution. Reports regarding dendrigraft polymers say that they are elongated due to linear polymeric core with dendritic arms. But in solution they remain as spherical.54
 | ||
| Fig. 11 TEM image of PEN-G2 polymer. | ||
2.3 Characterization of chiral modified dendrigraft PEN-G2 metal complex
The synthesis of the resin supported chiral modified dendrigraft PEN-Gn copper complex was achieved by adopting a three-step methodology, as shown in Scheme 5. Chiral modification of PEN-G2 polymer was done using Boc-L-proline followed by deprotection using trifluoroacetic acid. After chiral modification, amount of proline was found to be 12.54 and 20.63 mmol g−1 for G1 and G2 respectively. Chiral modified dendrigraft PEN-G2 polymer was complexed with copper using copper acetate in methanol at a temperature of about 50 °C. Chiral PEN-Gn–Cu was found to be non-hygroscopic, stable and can be stored for a prolonged period of time without any change in its catalytic efficiency. The copper coordinated dendritic polymer appeared as light green powder.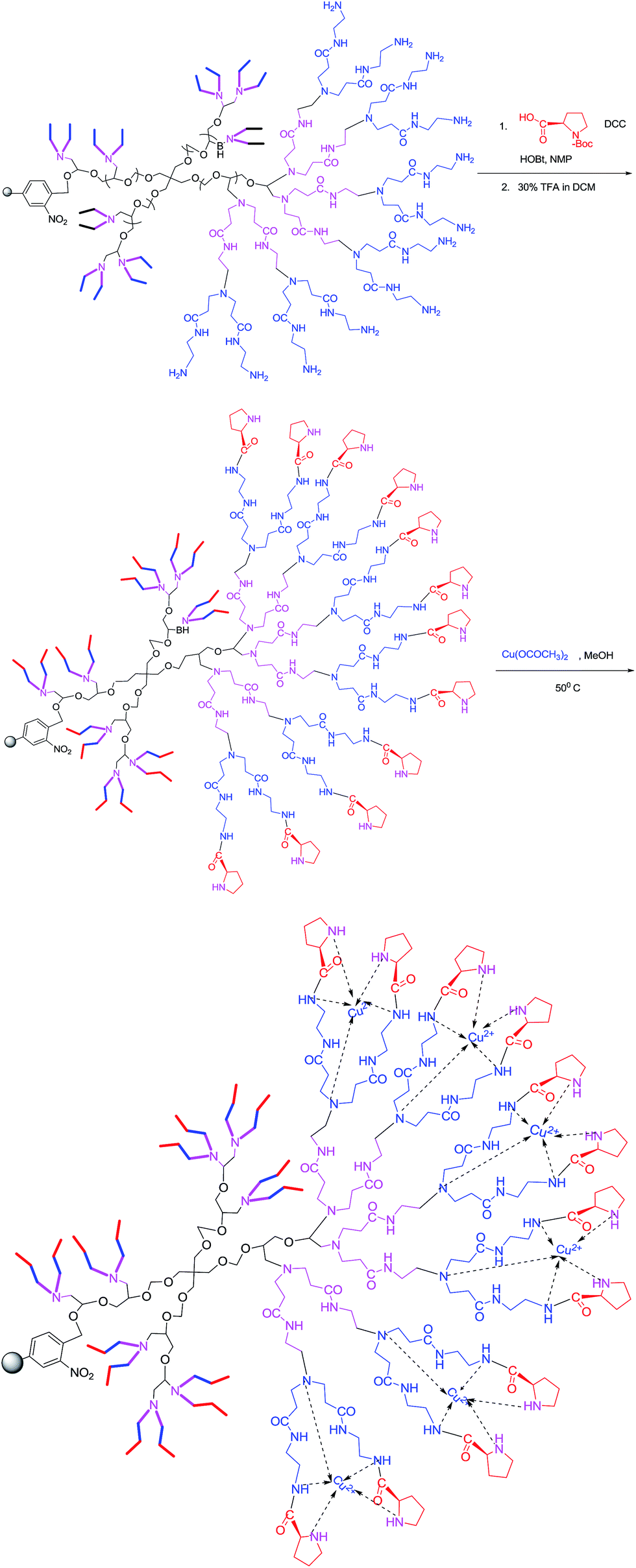 | ||
| Scheme 5 Chiral modification and metal complexation of PEN-G2 polymer. | ||
| Polymer | Proline NH capacity (mmol g−1) | Copper (%) ICP-AES | Copper (mmol g−1) ICP-AES | Copper (%) EDX |
|---|---|---|---|---|
| Chiral PEN-G1–(Cu) | 12.54 | 20.40 | 3.21 | 19.23 |
| Chiral PEN-G2–(Cu) | 20.63 | 32.36 | 5.09 | 30.27 |
 | ||
| Fig. 12 Scanning electron micrograph of chiral PEN-G2–Cu. | ||
 | ||
| Fig. 13 IR spectra comparison for chiral modification. | ||
Proline modified polymer on complexation with copper showed a distinct shift towards lower frequency region. Stretching due to proline NH was lowered around 64 cm−1 and that of amide carbonyl stretching was lowered from 1692 cm−1 to 1625 cm−1. The peak at 1508 cm−1 is due to stretching vibrations of secondary amine. Bending vibrations due to primary amine at 1610 cm−1 in the PEN-G2 polymer got modified by the secondary NH vibrations in chiral PEN-G2 polymer. The peak at 1200 cm−1 is due to the CH2 wagging of cyclopentane ring. After chiral modification, the peak at 1007 cm−1 in PEN-G2 due to the ethereal C–O stretching got merged with nearby peaks and shifted towards higher frequency region. The narrow peak at 700 cm−1 can be assigned to Cu–N stretching.
| Polymer | g∥ | g⊥ | gav | A∥ | A⊥ | Aav |
|---|---|---|---|---|---|---|
| PEN-G2–Cu | 2.28 | 2.07 | 2.14 | 195 | 12.8 | 69.83 |
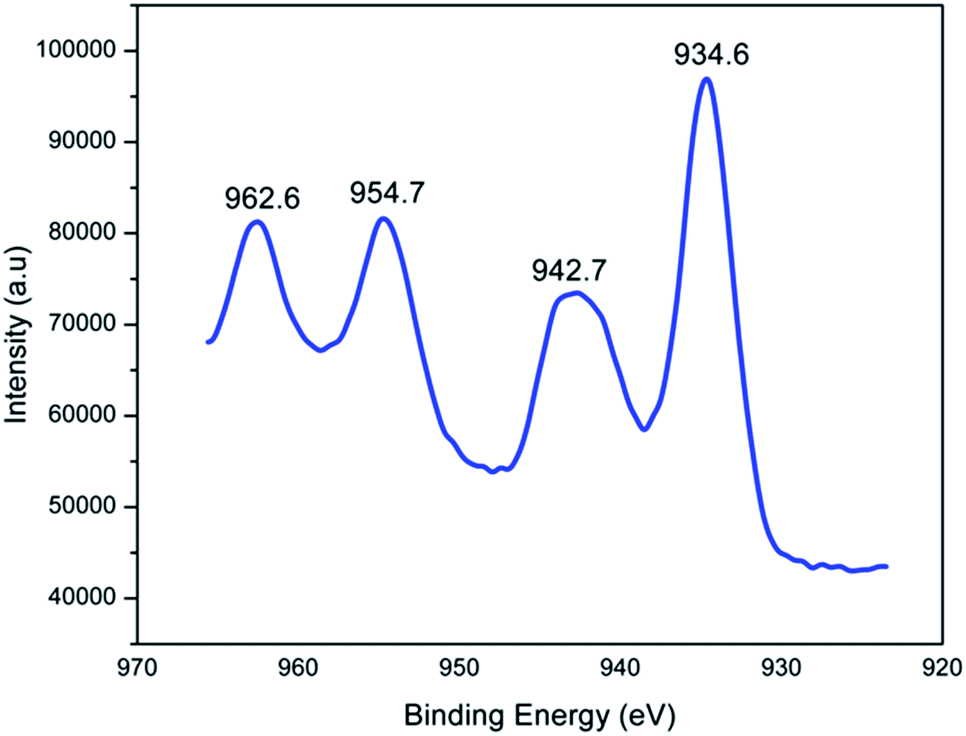 | ||
| Fig. 14 Deconvoluted XPS spectra of Cu (2p3/2) of PEN-G2–Cu. | ||
 | ||
| Fig. 15 TG-DTG plot of PEN-G2–Cu. | ||
Apart from this, in the case of PEN-G2–Cu, a decomposition step with a weight loss of 25% occurs in the temperature range of 150–250 °C. The thermal decomposition of PEN-G2 shows a weight loss of 30% which occurs in the temperature range of 120–320 °C. From the reports regarding polyepichlorohydrin and Merrifield resin,59 this decomposition is ascribed to the release of amines as nitrogen, carbonyls as CO or CO2 and the degradation of the polymeric backbone to be left with a residue containing copper oxide or hydroxide. The first step of decomposition in chiral PEN-G2–Cu is attributable to the loss of non-coordinated water, occurring in the temperature range of 65–105 °C. The TG-DTG analysis data for the dendrigraft polymers are thus in agreement with the percentage composition.
2.4 Catalytic activity of supported chiral dendrigraft PEN-Gn–Cu complex
 | ||
| Scheme 6 Aza Diels–Alder reaction of cyclohexenone with aldimines. | ||
It was found that chiral PEN-G2–Cu catalyst was more efficient than chiral PEN-G1–Cu catalyst. A variety of aldehydes as substrates has been tried. The effects of various reaction parameters, such as type of solvent, reaction temperature, catalyst concentration, etc., were evaluated using benzaldehyde, aniline and cyclohexenone as model substrates and chiral PEN-G2–Cu as the catalyst. Optimization studies of solvent and catalyst concentration are shown in Tables 3 and 4.
| Entry | Solvent | Yieldb (%) | (Endo/exo)c | eed (%) |
|---|---|---|---|---|
a Reaction conditions: cyclohexenone (1 mmol), benzaldehyde (1 mmol), aniline (1 mmol). RT, PEN-G2–Cu: 20 mg.b Isolated yield of endo and exo.c Determined by 1H NMR.d Enantiomeric excess of the major product was determined by chiral HPLC analysis (Chiralpak IB-3 chiral stationary phase, iPrOH/hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9), 254 nm). :9), 254 nm). |
||||
| 1 | DMSO | 60 | 82/18 | 80 |
| 2 | CCl4 | 82 | 92/08 | 90 |
| 3 | Ethanol | 78 | 76/24 | 74 |
| 4 | Hexane | 72 | 50/50 | 64 |
| 5 | Toluene | 82 | 60/40 | 75 |
| 6 | THF | 78 | 55/45 | 78 |
| 7 | CH3CN | 82 | 75/25 | 74 |
| Catalyst amount (mg) | Catalyst amount (mol%) | Yieldb (%) | TON, TOF (h−1) | (Endo/exo)c | eed (%) |
|---|---|---|---|---|---|
| a Reaction conditions: cyclohexenone (1 mmol), benzaldehyde (1 mmol), aniline (1 mmol). CCl4-3 mL, RT.b Isolated yield of endo and exo.c Determined by 1H NMR.d Enantiomeric excess of the major product was determined by chiral HPLC analysis (Chiralpak IB-3 chiral stationary phase, iPrOH/hexane (1:9), 254 nm). |
|||||
| 2.0 | 0.0010 | 53 | 53, 10.6 | 60/40 | 72 |
| 7.0 | 0.0036 | 65 | 18, 3.6 | 75/25 | 74 |
| 10.0 | 0.0051 | 82 | 16, 3.2 | 92/08 | 90 |
| 15.0 | 0.0076 | 82 | 11, 2.2 | 88/12 | 78 |
| 20.0 | 0.0102 | 82 | 8, 1.6 | 90/10 | 90 |
The reaction of cyclohexenone with imine was carried out in several solvents at an ambient temperature of 30 °C for 5 h. The results are summarized in Table 3. As shown in the table, selected solvents provide the product in reasonable yield with enantiomeric excess greater than 70%. CCl4 was chosen as the preferred solvent for further studies, because the best endo/exo ratio of the product was observed in this solvent. The efficiency of the catalyst was tested by varying the catalyst concentration.
As shown in Table 4, there was no appreciable change in the enantiomeric excess of the major product while decreasing the catalyst loading from 0.01 to 0.005 mol%. However, the best yield and % ee were achieved at 0.0051 mol% of the catalyst. We have also checked the influence of the amount of solvent of the reaction mixture on the catalyst efficiency. A dilution led to a decrease of the yield but the best % ee of the product was achieved with 3 mL of carbon tetrachloride.
Having established the optimum reaction conditions, effect of generation of the dendrigraft PEN-Gn–Cu was studied (Table 5). A positive dendritic effect was observed as the % ee increased with generation of the polymer. Indeed, first generation resulted in endo/exo ratio of 75/25, the second generation furnished endo/exo product ratio with 92/08 with 90% ee.
| Polymer | Yieldb (%) | TON, TOF (h−1) | (Endo/exo)c | eed (%) |
|---|---|---|---|---|
| a Reaction conditions: cyclohexenone (1 mmol), benzaldehyde (1 mmol), aniline (1 mmol). CCl4-3 mL, RT, catalyst amount: 10 mg.b Isolated yield of endo and exo.c Determined by 1H NMR.d Enantiomeric excess of the major product was determined by chiral HPLC analysis (Chiralpak IB-3 chiral stationary phase, iPrOH/hexane (1:9), 254 nm). |
||||
| PEN-G1–Cu | 80 | 25, 5.0 | 75/25 | 85 |
| PEN-G2–Cu | 82 | 16, 3.2 | 92/08 | 90 |
After optimizing the reaction conditions, scope of different substrates was evaluated (Table 6). All the substrates selected were studied for 3 h. Most of the reactions of cyclohexenone with imines resulted in good diastereo and enantioselectivity irrespective of the functional group on the aromatic ring. 1H NMR and GC-MS spectra of few diastereomers are shown in supplementary information. The diastereomeric ratio can be found out from proton NMR spectrum as well as from the GC-MS. In GC-MS, splitting pattern corresponding to endo and exo will be different which helps to predict diastereomeric ratio.
| Aza Diels–Alder product | Optimized structure | Yieldb (%) | Endo/exoc | eed (%) |
|---|---|---|---|---|
| a Reaction conditions: cyclohexenone (1 mmol), aldehyde (1 mmol), amine (1 mmol). CCl4-3 mL, RT, catalyst amount: 5.1 mol%, 3 h.b Isolated yield of endo and exo.c Determined by 1H NMR.d Enantiomeric excess of the major product was determined by chiral HPLC analysis (Chiralpak IB-3 chiral stationary phase, iPrOH/hexane (1:9), 254 nm). |
||||
 |
 |
82 | 92/08 | 90 |
 |
 |
82 | 88/12 | 84 |
 |
 |
84 | 87/13 | 86 |
 |
 |
62 | 84/16 | 78 |
 |
 |
82 | 88/12 | 88 |
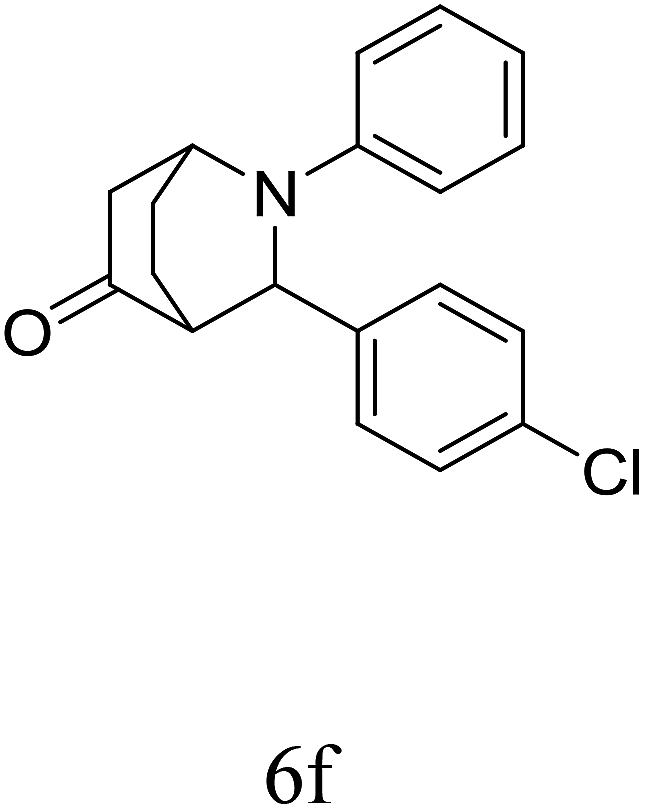 |
 |
84 | 80/20 | 88 |
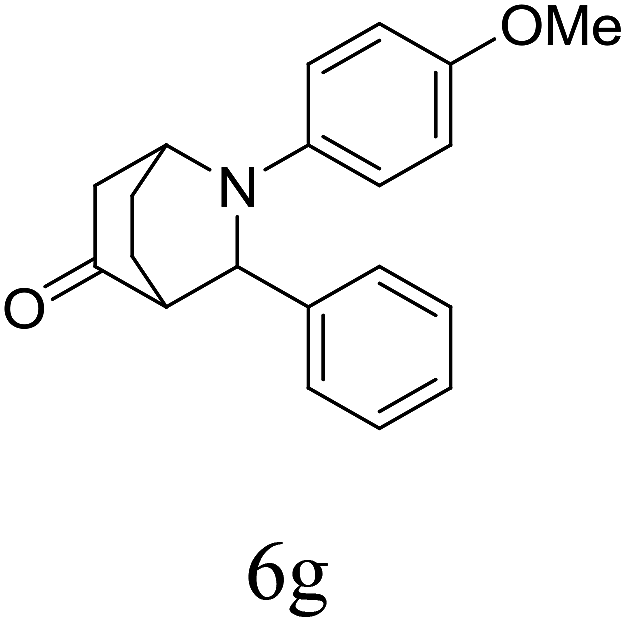 |
 |
85 | 88/12 | 90 |
 |
 |
76 | 90/10 | 86 |
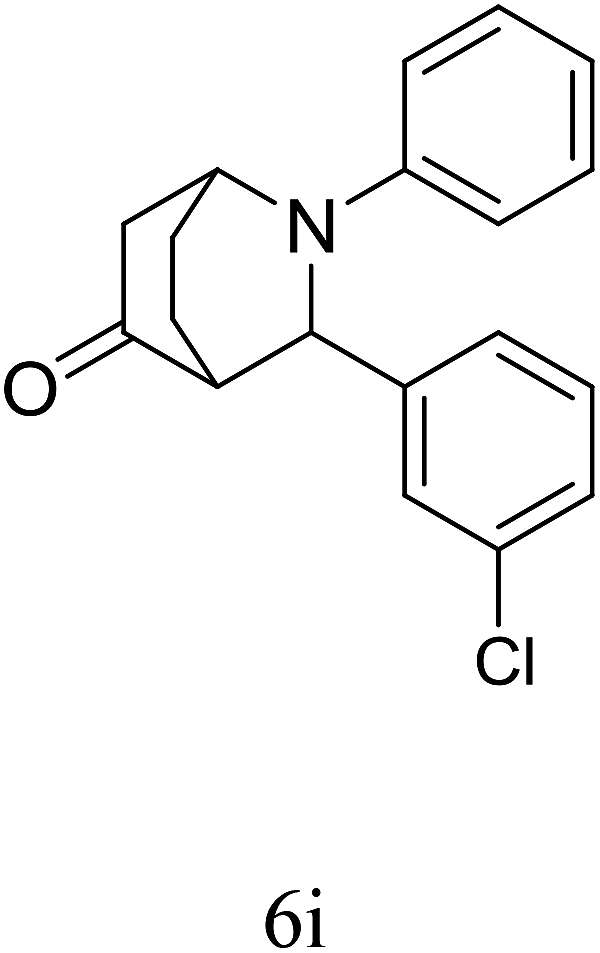 |
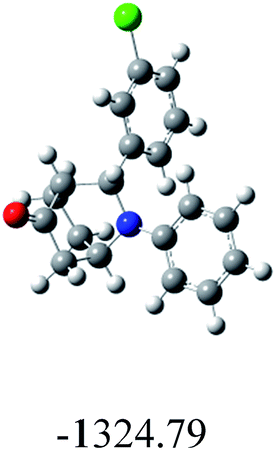 |
89 | 90/10 | 90 |
 |
 |
68 | 60/40 | 88 |
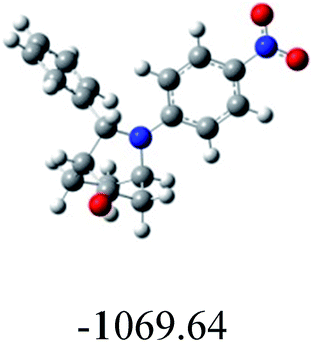
In order to find whether the yield of the aza Diels Alder product depends on its ground state energy, structure of the products were optimized using B3LYP exchange correlation functional and 6-31G basis set.60 The optimized energy i.e. lowest energy of the compound to be in the stable state, is depicted in the table (Table 6). As per the energy values, the compound 6a has maximum energy; so we expect it to have low yield (endo product) compared with others. But experimental results show endo/exo ratio of 92:08. In some other cases, the experimental values are agreeable with the theoretical value. For example, compound 6h have least energy value (−3550.67) which shows yield of about 90:10. These results show that formation of aza Diels–Alder product cannot be predicted by ground state energy values alone, but other factors (kinetic and thermodynamic factors) have to be considered in order to predict the formation of the product theoretically.
| No. of cycles | Catalyst weight (mg) | Catalyst recovered (mg) | Recovery (%) | Product yieldb (%) | Endo/exoc | eed (%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: cyclohexenone (1 mmol), benzaldehyde (1 mmol), aniline (1 mmol). CCl4-3 mL, RT, 3 h.b Isolated yield of endo and exo.c Determined by 1H NMR.d Enantiomeric excess of the major product was determined by chiral HPLC analysis (Chiralpak IB-3 chiral stationary phase, iPrOH/hexane (1:9), 254 nm). |
||||||
| 1 | 10 | 9.2 | 92.0 | 82 | 92/08 | 90 |
| 2 | 9.2 | 8.4 | 91.3 | 78 | 82/18 | 82 |
| 3 | 8.4 | 7.8 | 92.9 | 74 | 78/22 | 78 |
| 4 | 7.8 | 7.4 | 94.9 | 65 | 75/25 | 74 |
| 5 | 7.4 | 6.5 | 87.8 | 65 | 74/26 | 74 |
The (4 + 2) diene–dienophile cycloaddition of enolate with aldimine generates intermediate I and II. Finally catalyst leaves the system to form the product, endo and exo (Scheme 7).
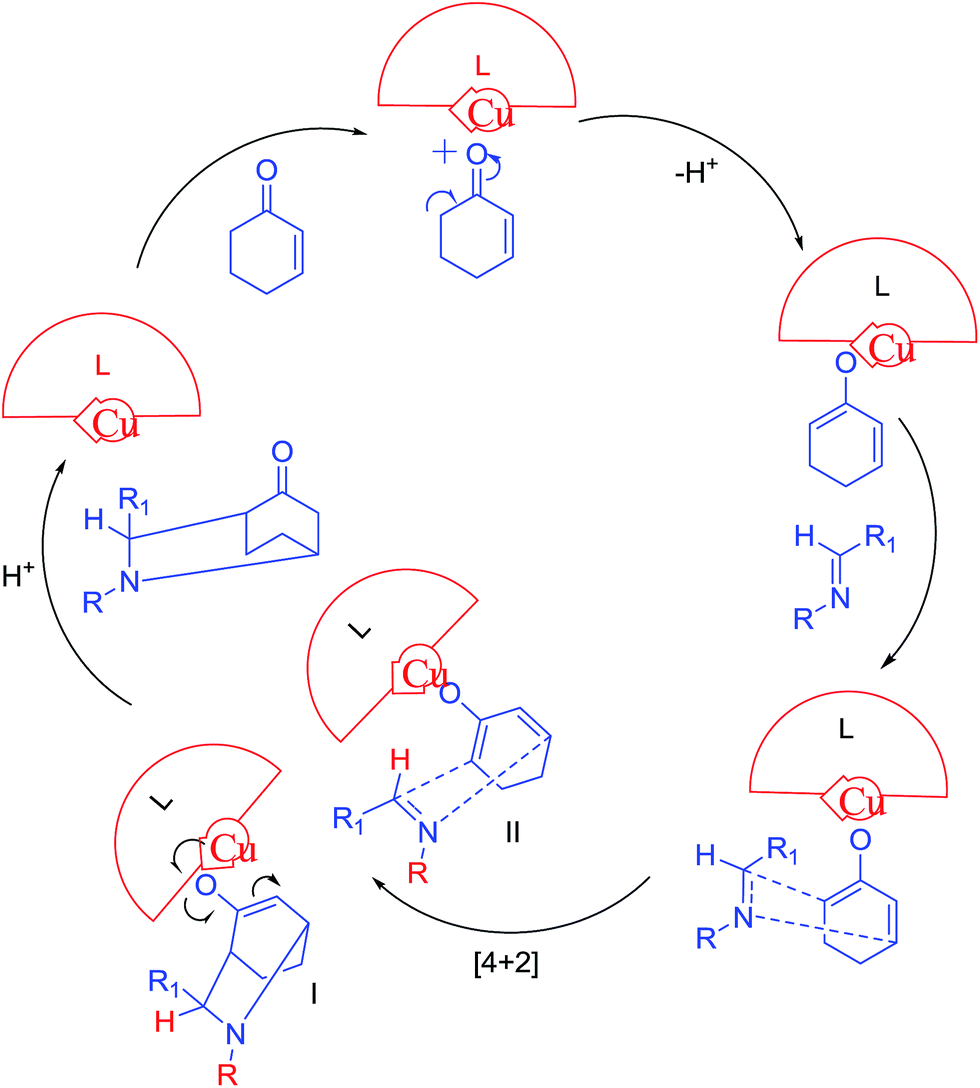 | ||
| Scheme 7 Possible mechanism for aza Diels Alder reaction of cyclohexenone with aldimines. | ||
3. Experimental
3.1 Photolytic cleavage
Resin (500 mg) was suspended in methanol (50 mL) in the reaction chamber of an immersion type photo reactor. The suspension was degassed for 1 h with dry nitrogen and irradiated with Philips HP 125 W medium pressure Hg lamp at 340–350 nm for 24 h with constant stirring. A solution of CuSO4 was circulated through the outer jacket of the reactor to filter off light waves below 320 nm. After photolysis, resin beads were filtered and washed with methanol. Combined filtrate and washings were evaporated under vacuum.3.2 Modification of dendrigraft PEN-G2 polymer with Boc proline
Active ester of amino acid was prepared by adding 2.5 meq. of HOBt (1.69 g) and 2.5 meq. of DCC (2.58 g) to a solution of Boc-L-proline (2.69 g) in NMP. This was stirred for 5 min and the HOBt ester of amino acid was added to the amino resin, PEN-G2 (0.5 g). The mixture was shaken for 48 h until the disappearance of blue colour with ninhydrin. DMSO was added to the mixture and shaken for 15 min. At the end of 15 min. DIEA was added. The unreacted reagents and byproducts were filtered off. The resin was washed with dichloromethane:methanol (66:33 v/v), dichloromethane, NMP and dried under vacuum at 50 °C.
Amount of Boc-proline: 0.021 mol g−1; yield: 0.58 g.
IR (cm−1): 3460, 2934, 1740, 1652, 1539, 1366, 1103, 725.
Solid state 13C NMR (100 MHz): 172, 168.2, 167.8, 167.2, 152, 144.3, 122.2, 79.5, 76, 73, 61, 60.3, 47, 43, 38.8, 34, 29, 27, 23, 20.9 ppm.
3.3 Deprotection of Boc from dendrigraft PEN-G2 polymer
The resin (0.5 g) was treated with 30% TFA in dichloromethane for 5 h. The reaction was monitored using ninhydrin test and IR spectrum. The TFA solution was filtered and the resin was washed with dichloromethane. This was treated with 5% DIEA in dichloromethane (5 min) & 5% DIEA in NMP:dichloromethane mixture (1:1 v/v) to get the desired product. Finally, proline modified PEN-G2 polymer was dried at 50 °C.
Proline NH capacity: 0.021 mol g−1; yield: 0.46 g; IR (cm−1): 3460, 2982, 1692, 1525, 1454, 1250, 1117, 750.
Solid state 13C NMR (100 MHz): 168, 167.5, 167, 142.3, 122.4, 79.9, 73, 64, 62.3, 49, 42, 37, 35, 30, 25, 20.2 ppm.
3.4 Synthesis of copper complex of proline modified dendrigraft PEN-G2 polymer
A 50 mL round bottom flask was charged with 250 mg of Merrifield resin supported proline modified PEN-G2 polymer having 20.63 mmol g−1 of amine capacity. It was allowed to swell in DMF for 2 h. Quantitative amount of anhydrous copper acetate (0.021 mol, 0.94 g) in 10 mL methanol was added to the reaction flask. The reaction mixture was stirred at 50 °C for about two days (48 h). The polymer was filtered and washed with water. The filtrate and washings were collected together and concentrated. This concentrated solution was used for the estimation of metal ions by standard methods. The polymer-supported metal complex was washed with methanol (20 mL × 3), dioxane (20 mL × 3) and acetone (20 mL × 3) followed by drying at 50 °C for 3 h.Dark green powder; copper loading: 0.005 mol g−1; yield: 0.32 g.
3.5 General procedure for aza Diels Alder reaction
A 25 mL RB flask was charged with a magnetic stirrer and CCl4 (3.0 mL). Cyclohexenone (1.0 mmol), and the catalyst PEN-G2–Cu (0.0051 mol%, 10 mg) were added to the flask. To the resulting solution after 2 min of stirring, the Schiff base of aniline and benzaldehyde (1 mmol) was added. The reaction mixture was stirred at room temperature for 3 h. After the completion of the reaction (TLC and GC determination), the resulting solution was filtered, washed with ethyl acetate, methanol and acetone, and concentrated by a rotary vacuum evaporator. The product was separated by passing through a silica column using the eluent, hexane:ethyl acetate (9:1). The yield and diastereomer ratio were determined from 1H NMR spectrum. Product purification and diastereomer separation were made by column chromatography on SiO2 column (hexane/EtOAc/Et3N 40:10:1). The enantiomeric purity was determined by chiral HPLC analysis (Chiralpak IB-3 chiral stationary phase, iPrOH/hexane (1:9), 254 nm).
4. Conclusions
Polyepichlorohydrin was coupled to Merrifield resin in order to increase the loading capacity. Novel families of dendrigraft G0, G1 and G2 amine polymer having pentaerythritol initiated polyepichlorohydrin as core have been synthesized and characterized. Characterization of dendrigraft amidoamine polymer has been done after photolytic cleavage of the same from the support. We have developed a highly efficient chiral dendritic copper catalyzed method for the cycloaddition of cyclohexenone with aldimines. The developed copper complexes of chiral PEN-G1 and PEN-G2 dendrigraft polymer were characterized. We found that copper complexes can catalyze aza Diels–Alder reaction like Diels–Alder reaction. The reaction occurs more efficiently with high generation dendrigraft polymer in comparison with low generation G1 dendrigraft polymer. After optimizing the reaction conditions, a detailed study of aza Diels–Alder reaction was done with chiral PEN-G2 copper catalyst. The main features of the synthesis are: only small amount of catalyst was needed to drive the reaction and all the reactions were performed at room temperature. The catalyst performs well with good diastereoselectivity and good enantiomeric excess. Procedural simplicity, simple recovery and reusability of catalysts meet the requirements of benign chemistry. Thus the present catalyst will be of wide practical application in similar reactions. Further investigation on the application of this catalyst for other organic reactions is in progress.Acknowledgements
We would like to thank SAIF, CUSAT for assistance with various analysis. One of the authors (G. S.) thank the CSIR, Govt. of India for financial assistance in the form of fellowship. The authors thank PSRT, CUSAT for GPC analysis, NCL, Pune for Solid state 13C NMR analysis, RGCB and NIIST, Trivandrum for MALDI analysis. Finally we thank Thermax, India for providing the gift sample of Merrifield resin.References
- (a) A. D. Schluter and J. P. Rabe, Angew. Chem., Int. Ed., 2000, 39, 864–883 CrossRef CAS; (b) A. D. Schluter, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1533–1556 CrossRef CAS; (c) A. F. Zhang, L. J. Shu, Z. S. Bo and A. D. Schluter, Macromol. Chem. Phys., 2003, 204, 328–339 CrossRef CAS; (d) H. Frauenrath, Prog. Polym. Sci., 2005, 30, 325–384 CrossRef CAS; (e) C. C. Lee, J. A. MacKay, J. M. J. Frechet and F. C. Szoka, Nat. Biotechnol., 2005, 23, 1517–1526 CrossRef CAS PubMed; (f) J. G. Rudick and V. Percec, Acc. Chem. Res., 2008, 41, 1641–1652 CrossRef CAS PubMed; (g) B. M. Rosen, C. J. Wilson, D. A. Wilson, M. Peterca, M. R. Imam and V. Percec, Chem. Rev., 2009, 109, 6275–6540 CrossRef CAS PubMed; (h) W. Li, A. Zhang and A. D. Schlueter, Macromolecules, 2008, 41, 43–49 CrossRef CAS; (i) W. Li, A. Zhang, K. Feldman, P. Walde and A. D. Schlueter, Macromolecules, 2008, 41, 3659–3667 CrossRef CAS; (j) W. Zhuang, E. Kasemi, Y. Ding, M. Kroeger, A. D. Schlueter and J. P. Rabe, Adv. Mater., 2008, 20, 3204–3210 CrossRef CAS; (k) A. J. Boydston, T. W. Holcombe, D. A. Unruh, J. M. J. Frechet and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 5388–5389 CrossRef CAS PubMed; (l) Y. Guo, J. D. van Beek, B. Zhang, M. Colussi, P. Walde, A. Zhang, M. Kroeger, A. Halperin and A. D. Schlueter, J. Am. Chem. Soc., 2009, 131, 11841–11854 CrossRef CAS PubMed; (m) I. Popa, B. Zhang, P. Maroni, A. D. Schlueter and M. Borkovec, Angew. Chem., Int. Ed., 2010, 49, 4250–4253 CrossRef CAS PubMed; (n) B. Zhang, R. Wepf, K. Fischer, M. Schmidt, S. Besse, P. Lindner, B. T. King, R. Sigel, P. Schurtenberger, Y. Talmon, Y. Ding, M. Kroeger, A. Halperin and A. D. Schlueter, Angew. Chem., Int. Ed., 2011, 50, 737–740 CrossRef CAS PubMed; (o) R. M. England and S. Rimmer, Polym. Chem., 2010, 1, 1533–1544 RSC; (p) Y. Deng, S. Zhang, G. Lu and X. Huang, Polym. Chem., 2013, 4, 1289–1299 RSC; (q) C. Li, H. Liu, D. Tang and Y. Zhao, Polym. Chem., 2015, 6, 1474–1486 RSC; (r) C. Guo, L. Sun, W. She, N. Li, L. Jiang, K. Luo, Q. Gong and Z. Gu, Polym. Chem., 2016, 7, 2531–2541 RSC.
- J. S. Fruchtel and G. Jung, Angew. Chem., Int. Ed., 1996, 35, 17–42 CrossRef.
- G. Smitha and K. Sreekumar, RSC Adv., 2016, 6, 18141–18155 RSC.
- (a) R. Haag, Chem.–Eur. J., 2001, 7, 327–335 CrossRef CAS PubMed; (b) S. Lebreton, S. Monaghan and M. Bradley, Aldrichimica Acta, 2001, 34, 75–83 CAS.
- V. Swali, N. J. Wells, G. J. Langley and M. Bradley, J. Org. Chem., 1997, 62, 4902–4903 CrossRef CAS.
- D. A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Polym. J., 1985, 17, 117 CrossRef CAS.
- N. J. Wells, A. Basso and M. Bradley, Biopolymers, 1998, 47, 381–396 CrossRef CAS.
- N. J. Wells, M. Davies and M. Bradley, J. Org. Chem., 1998, 63, 6430–6431 CrossRef CAS.
- (a) A. Dahan and M. Portnoy, J. Am. Chem. Soc., 2007, 129, 5860–5869 CrossRef CAS PubMed; (b) T. Kehat, K. Goren and M. Portnoy, New J. Chem., 2007, 31, 1218–1242 RSC; (c) A. Y.-T. Huang, C.-H. Tsai, H.-Y. Chen, H.-T. Chen, C.-Y. Lu, Y.-T. Lin and C.-L. Kao, Chem. Commun., 2013, 49, 5784–5786 RSC; (d) R. K. G. Panicker and S. Krishnapillai, Tetrahedron Lett., 2014, 55, 2352–2354 CrossRef; (e) P. Arya, N. V. Rao, J. Singkhonrat, H. Alper, S. C. Bourque and L. E. Manzer, J. Org. Chem., 2000, 65, 1881–1885 CrossRef CAS PubMed; (f) S. Lebreton, N. Newcombe and M. Bradley, Tetrahedron Lett., 2002, 43, 2475–2478 CrossRef CAS; (g) J. Lu and P. H. Toy, Chem. Rev., 2009, 109, 815–838 CrossRef CAS PubMed; (h) E. Delort, N.-Q. Nguyen-Trung, T. Darbre and J.-L. Reymond, J. Org. Chem., 2006, 71, 4468–4480 CrossRef CAS PubMed; (i) J. J. Diaz-Mochon, M. A. Fara, R. M. Sanchez-Martin and M. Bradley, Tetrahedron Lett., 2008, 49, 923–926 CrossRef CAS; (j) K. Goren and M. Portnoy, Chem. Commun., 2010, 46, 1965–1967 RSC; (k) T. Kehat and M. Portnoy, Chem. Commun., 2007, 2823–2825 RSC; (l) L. Tuchman-Shukron and M. Portnoy, Adv. Synth. Catal., 2009, 351, 541–546 CrossRef CAS; (m) M. P. Kapoor, Y. Kasama, T. Yokoyama, M. Yanagi, S. Inagaki, N. Hironobu and L. R. Juneja, J. Mater. Chem., 2006, 16, 4714–4722 RSC; (n) G. R. Krishnan and K. Sreekumar, Eur. J. Org. Chem., 2008, 4763–4768 CrossRef CAS; (o) J.-P. Lindner, C. Roeben, A. Studer, M. Stasiak, R. Ronge, A. Greiner and H.-J. Wendorff, Angew. Chem., Int. Ed., 2009, 48, 8874–8877 CrossRef CAS PubMed; (p) G. R. Krishnan and K. Sreekumar, Polymer, 2008, 49, 5233–5240 CrossRef CAS; (q) A.-M. Caminade, A. Ouali, M. Keller and J.-P. Majoral, Chem. Soc. Rev., 2012, 41, 4113–4125 RSC.
- (a) G. Consiglio, in Catalytic Asymmetric Synthesis, ed. I. Ojima, VCH, New York, 1993 Search PubMed; (b) R. Noyori, Asymmetric Catalysis in Organic Synthesis, John Wiley & Sons, New York, 1994 Search PubMed; (c) M. Rueping, B. J. Nachtsheim, W. Ieawsuwan and I. Atodiresei, Angew. Chem., Int. Ed., 2011, 50, 6706–6720 CrossRef CAS PubMed; (d) V. Eschenbrenner-Lux, K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2014, 53, 11146–11157 CrossRef CAS PubMed; (e) Z. Y. Cao, W. D. G. Brittain, J. S. Fossey and F. Zhou, Catal. Sci. Technol., 2015, 5, 3441–3451 RSC.
- (a) M. G. Memeo and P. Quadrelli, Chem.–Eur. J., 2012, 18, 12554–12582 CrossRef CAS PubMed; (b) S. Kobayashi and H. Ishitani, Chem. Rev., 1999, 99, 1069–1094 CrossRef CAS PubMed; (c) H. B. Kagan and O. Riant, Chem. Rev., 1992, 92, 1007–1019 CrossRef CAS; (d) K. A. Jorgensen, Angew. Chem., Int. Ed., 2000, 39, 3558–3588 CrossRef CAS.
- (a) A. D. Schluter, in Functional Molecular Nanostructures, Top. Curr. Chem., 2005, vol. 245, pp. 151–191 Search PubMed; (b) K. Ishizu, K. Tsubaki, A. Mori and S. Uchida, Prog. Polym. Sci., 2003, 28, 27–54 CrossRef CAS.
- (a) T. Akiyama, J. Itoh and K. Fuchibe, Adv. Synth. Catal., 2006, 348, 999–1010 CrossRef CAS; (b) J. Itoh, K. Fuchibe and T. Akiyama, Angew. Chem., Int. Ed., 2006, 45, 4796–4798 CrossRef CAS PubMed; (c) U. K. Tambar, S. K. Lee and J. L. Leighton, J. Am. Chem. Soc., 2010, 132, 10248–10250 CrossRef CAS PubMed; (d) S. Kobayashi, S. Komiyama and H. Ishitani, Angew. Chem., Int. Ed., 1998, 37, 979–981 CrossRef CAS; (e) S. Kobayashi, K. Kusakabe and H. Ishitani, Org. Lett., 2000, 2, 1225–1227 CrossRef CAS PubMed; (f) S. Kobayashi, K. Kusakabe, S. Komiyama and H. Ishitani, J. Org. Chem., 1999, 64, 4220–4221 CrossRef CAS; (g) Y. Yamashita, Y. Mizuki and S. Kobayashi, Tetrahedron Lett., 2005, 46, 1803–1806 CrossRef CAS; (h) N. S. Josephsohn, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2003, 125, 4018–4019 CrossRef CAS PubMed; (i) O. G. Mancheno, R. G. Arrayas and J. C. Carretero, J. Am. Chem. Soc., 2004, 126, 456–457 CrossRef CAS PubMed; (j) S. L. Yao, S. Saaby, R. G. Hazell and K. A. Jorgensen, Chem.–Eur. J., 2000, 6, 2435–2448 CrossRef CAS PubMed; (k) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520–1543 CrossRef CAS PubMed; (l) Y. Takemoto, Org. Biomol. Chem., 2005, 3, 4299–4306 RSC; (m) A. K. Unni, N. Takenaka, H. Yamamoto and V. H. Rawal, J. Am. Chem. Soc., 2005, 127, 1336–1337 CrossRef CAS PubMed.
- (a) D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356–5357 CrossRef CAS PubMed; (b) T. Akiyama, H. Morita, J. Itoh and K. Fuchibe, Org. Lett., 2005, 7, 2583–2585 CrossRef CAS PubMed; (c) R. I. Storer, D. E. Carrera, Y. Ni and D. W. C. MacMillan, J. Am. Chem. Soc., 2006, 128, 84–86 CrossRef CAS PubMed; (d) J. Seayad, A. M. Seayad and B. List, J. Am. Chem. Soc., 2006, 128, 1086–1087 CrossRef CAS PubMed; (e) D. Uraguchi, K. Sorimachi and M. Terada, J. Am. Chem. Soc., 2005, 127, 9360–9361 CrossRef CAS PubMed; (f) M. Terada, K. Machioka and K. Sorimachi, Angew. Chem., Int. Ed., 2006, 45, 2254–2257 CrossRef CAS PubMed; (g) M. Rueping, A. R. Antonchick and T. Theissmann, Angew. Chem., Int. Ed., 2006, 45, 2617–2619 CrossRef PubMed; (h) S. Mayer and B. List, Angew. Chem., Int. Ed., 2006, 45, 4193–4195 CrossRef CAS PubMed; (i) X.-H. Chen, X.-Y. Xu, H. Liu, L.-F. Cun and L.-Z. Gong, J. Am. Chem. Soc., 2006, 128, 14802–14803 CrossRef CAS PubMed.
- H. Ishitani and S. Kobayashi, Tetrahedron Lett., 1996, 37, 7357–7360 CrossRef CAS.
- (a) H. Ishitani, M. Ueno and S. Kobayashi, J. Am. Chem. Soc., 1997, 119, 7153–7154 CrossRef; (b) K. Hattori and H. Yamamoto, Tetrahedron, 1993, 49, 1749–1760 CrossRef CAS.
- S. K. Lee, U. K. Tambar, N. R. Perl and J. L. Leighton, Tetrahedron, 2010, 66, 4769–4774 CrossRef CAS.
- W. Zhang, Y. Dai, X. Wang and W. Zhang, Tetrahedron Lett., 2011, 52, 6122–6126 CrossRef CAS.
- Z. Chen, L. Lin, D. Chen, J. Li, X. Liu and X. Feng, Tetrahedron Lett., 2010, 51, 3088–3091 CrossRef CAS.
- H. Shirakawa and H. Sano, Tetrahedron Lett., 2014, 55, 4095–4097 CrossRef CAS.
- H. Sunden, I. Ibrahem, L. Eriksson and A. Cordova, Angew. Chem., Int. Ed., 2005, 44, 4877–4880 CrossRef CAS PubMed.
- H. Liu, L.-F. Cun, A.-Q. Mi, Y.-Z. Jiang and L.-Z. Gong, Org. Lett., 2006, 8, 6023–6026 CrossRef CAS PubMed.
- V. I. Maleev, T. y. V. Skrupskaya, L. V. Yashkina, A. F. Mkrtchyan, A. S. Saghyan, M. M. Il'in and D. A. Chusov, Tetrahedron: Asymmetry, 2013, 24, 178–183 CrossRef CAS.
- H. Yamamoto, Lewis Acids in Organic Synthesis, Wiley-VCH, 2000 Search PubMed.
- F. M. Menger and T. Tsuno, J. Am. Chem. Soc., 1989, 111, 4903–4907 CrossRef CAS.
- (a) C. C. Mak and H. F. Chow, Macromolecules, 1997, 30, 1228–1230 CrossRef CAS; (b) H. F. Chow and C. C. Mak, J. Org. Chem., 1997, 62, 5116–5127 CrossRef CAS.
- K. I. Fujita, T. Muraki, H. Hattori and T. Sakakura, Tetrahedron Lett., 2006, 47, 4831–4834 CrossRef CAS.
- (a) L. He, G. Laurent, P. Retailleau, B. Folleas, J.-L. Brayer and G. Masson, Angew. Chem., Int. Ed., 2013, 52, 11088–11091 CrossRef CAS PubMed; (b) S. Reymond and J. Cossy, Chem. Rev., 2008, 108, 5359–5406 CrossRef CAS PubMed.
- (a) K. Juhl and K. A. Jorgensen, Angew. Chem., Int. Ed., 2003, 42, 1498–1501 CrossRef CAS PubMed; (b) Y. Yamamoto, N. Momiyama and H. Yamamoto, J. Am. Chem. Soc., 2004, 126, 5962–5963 CrossRef CAS PubMed; (c) Y. Hayashi, J. Yamaguchi, K. Hibino, T. Sumiya, T. Urushima, M. Shoji, D. Hashizume and H. Koshino, Adv. Synth. Catal., 2004, 346, 1435–1439 CrossRef CAS; (d) A. B. Northrup and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 2458–2460 CrossRef CAS PubMed; (e) D. B. Ramachary, N. S. Chowdari and C. F. Barbas, Angew. Chem., Int. Ed., 2003, 42, 4233–4237 CrossRef CAS PubMed; (f) K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243–4244 CrossRef CAS; (g) H. Sunden, N. Dahlin, I. Ibrahem, H. Adolfsson and A. Cordova, Tetrahedron Lett., 2005, 46, 3385–3389 CrossRef CAS.
- (a) J. Casas, M. Engqvist, I. Ibrahem, B. Kaynak and A. Cordova, Angew. Chem., Int. Ed., 2005, 44, 1343–1345 CrossRef CAS PubMed; (b) A. Cordova, H. Sunden, M. Engqvist, I. Ibrahem and J. Casas, J. Am. Chem. Soc., 2004, 126, 8914–8915 CrossRef CAS PubMed; (c) H. Sunden, M. Engqvist, J. Casas, I. Ibrahem and A. Cordova, Angew. Chem., Int. Ed., 2004, 43, 6532–6535 CrossRef CAS PubMed; (d) A. Cordova, Acc. Chem. Res., 2004, 37, 102–112 CrossRef CAS PubMed; (e) A. Bogevig, H. Sunden and A. Cordova, Angew. Chem., Int. Ed., 2004, 43, 1109–1112 CrossRef CAS PubMed; (f) A. Cordova, Chem.–Eur. J., 2004, 10, 1987–1997 CrossRef CAS PubMed; (g) A. Cordova, H. Sunden, A. Bogevig, M. Johansson and F. Himo, Chem.–Eur. J., 2004, 10, 3673–3684 CrossRef CAS PubMed.
- K. U. Ito, Polym. J., 1979, 11, 171–174 CrossRef CAS.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2151 CrossRef CAS.
- (a) A. Barbara, Tetrahedron Lett., 1973, 14, 21–25 CrossRef; (b) J. Karabline and M. Portnoy, Org. Biomol. Chem., 2012, 10, 4788–4794 RSC.
- S. K. Sahu, S. P. Panda, D. S. Sadafule, C. G. Kumbhar, S. G. Kulkarni and J. V. Thakur, Polym. Degrad. Stab., 1998, 62, 495–500 CrossRef CAS.
- B. Gaur, B. Lochab, V. Choudhary and I. K. Varma, J. Macromol. Sci., Polym. Rev., 2003, C43, 505–545 CrossRef CAS.
- H. J. Milton, E. C. Struck, M. G. Case and P. M. Steven, J. Polym. Sci., Polym. Chem. Ed., 1984, 22, 341–352 CrossRef.
- (a) T. Biedron, P. Kubisa and S. Penczek, J. Polym. Sci., Part A: Polym. Chem., 1991, 29, 619–628 CrossRef CAS; (b) G. Ampleman, US Pat., 5,124,463, 1992.
- P. Liu, Anal. Chim. Acta, 2005, 95, 695–698 CAS.
- H. Arisawa and T. B. Brill, Combust. Flame, 1998, 112, 533–544 CrossRef CAS.
- D. A. Tomaia, Macromolecules, 1986, 19, 2466–2472 CrossRef.
- F. Guillier, D. Orain and M. Bradley, Chem. Rev., 2000, 100, 2091–2157 CrossRef CAS PubMed.
- F. P. Luiz, R. Ricardo and F. M. Eduardo, J. Am. Chem. Soc., 2013, 135, 11513–11516 CrossRef PubMed.
- L. Shu, Dendronized Polystyrene: Synthesis, Characterization and SFM Investigations of Cylindrical Nano objects, PhD Dissertation, University of Berlin, Germany, 2001.
- (a) D. C. Schriemer and L. Li, Anal. Chem., 1997, 69, 4176–4183 CrossRef CAS; (b) A. M. Schaiberger and J. A. Moss, J. Am. Soc. Mass Spectrom., 2008, 19, 614–619 CrossRef CAS PubMed; (c) U. Bahr, A. Deppe, M. Karas, F. Hillenkamp and U. Giessmann, Anal. Chem., 1992, 64, 2866–2869 CrossRef CAS.
- D. A. Tomalia and J. M. J. Frechet, Dendrimers and Dendritic Polymers, Wiley series in Polymer Science, 2001, pp. 587–604 Search PubMed.
- S. S. Sheiko and M. Moller, Chem. Rev., 2001, 101, 4099–4123 CrossRef CAS PubMed.
- N. Thi-Thanh-Tam, B. Martin, R. Ali, J. R. Hans, I. Lieberwirth and K. Mullen, J. Am. Chem. Soc., 2013, 135, 4183–4186 CrossRef PubMed.
- M. F. Ottaviani, S. Bossmann, N. J. Turro and D. A. Tomalia, J. Am. Chem. Soc., 1994, 116, 661–671 CrossRef CAS.
- E.-H. Kang, I. S. Lee and T.-L. Choi, J. Am. Chem. Soc., 2011, 133, 11904–11907 CrossRef CAS PubMed.
- A. M. Naylor, W. A. Goddard, G. E. Kiefer and D. A. Tomaia, J. Am. Chem. Soc., 1989, 111, 2339–2341 CrossRef CAS.
- A. Carlmark, E. Malmstrom and M. Malkoch, Chem. Soc. Rev., 2013, 42, 5858–5879 RSC.
- J. Li, L. T. Piehler, D. Qin, J. R. Baker Jr and D. A. Tomalia, Langmuir, 2000, 16, 5613–5616 CrossRef CAS.
- L. J. Shu, T. Schafer and A. D. Schluter, Macromolecules, 2000, 33, 4321–4328 CrossRef CAS.
- D. Xie, M. Jiang, G. Zhang and D. Chen, Chem.–Eur. J., 2007, 13, 3346–3353 CrossRef CAS PubMed.
- L. Zhou, D. H. Russell, M. Q. Zhao and R. M. Crooks, Macromolecules, 2001, 34, 3567–3573 CrossRef CAS.
- (a) S. Thakurta, P. Roy, G. Rosair, C. J. Gomez-Garcia, E. Garribba and S. Mitra, Polyhedron, 2009, 28, 695–702 CrossRef CAS; (b) V. P. Singh, Spectrochim. Acta, Part A, 2008, 71, 17–19 CrossRef PubMed.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- S. K. Sahu, S. P. Panda, D. S. Sadafule, C. G. Kumbhar, S. G. Kulkarni and J. V. Thakur, Polym. Degrad. Stab., 1998, 62, 495–500 CrossRef CAS.
- S. Pisharath and H. G. Ang, Polym. Degrad. Stab., 2007, 92, 1365–1377 CrossRef CAS.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: GPC profile, IR, NMR, solid state 13C NMR, EDX and MALDI spectrum. SEM, TEM, AFM images. Experimental procedure, spectral data of products. See DOI: 10.1039/c6ra15548k |
| This journal is © The Royal Society of Chemistry 2016 |