
A numerical study of the photothermal behaviour of near-infrared plasmonic colloids†
Abstract
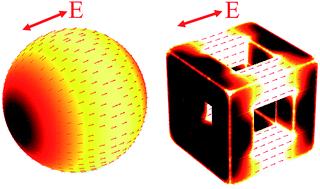
We study the photothermal properties of near-infrared absorbing colloidal plasmonic nanoparticles that are of interest for theranostic applications: SiO2@Au core–shell particles and Au nanocages. Three-dimensional (3D) optical and thermodynamic computational models are developed to explore and compare the optical and thermal response of these particles. The analysis demonstrates that plasmon-enhanced photothermal heating efficiency is a complex function of interrelated factors including the gold content of a particle and the degree to which it supports field-induced current to promote Joule heating. The thermal analysis elucidates fundamental mechanisms that govern the transient temperature distribution and heat dissipation produced by the particles. The modeling approach broadly applies to plasmonic nanostructures with arbitrary shapes, sizes and material constituents and is well suited for the rational design of novel plasmon-assisted photothermal applications.
 Please wait while we load your content...
Please wait while we load your content...

