
Fabrication and characterization of polymer–ceramic nanocomposites containing pluronic F127 immobilized on hydroxyapatite nanoparticles†
Mohammad Masoud Mirhosseini,
Vahid Haddadi-Asl* and
Seyed Shahrooz Zargarian
Department of Polymer Engineering and Color Technology, Amirkabir University of Technology, Tehran, Iran. E-mail: haddadi@aut.ac.ir; Tel: +98 21 64542305
First published on 17th August 2016
Abstract
Functionalized hydroxyapatite nanoparticles (HA-F127) were synthesized by immobilizing pluronic F127 onto hydroxyapatite nanoparticles (HA). Successful grafting of F127 was evaluated by FTIR, TGA and TEM. XRD was used to study the crystalline structure of neat and functionalized nanoparticles. Grafted F127 chains on the surface of HA formed a core–shell structure. Furthermore, immobilization of F127 chains on the HA surface decreased the agglomeration of modified nanoparticles and improved their dispersion. In the next step, the HA-F127 and unmodified HA were introduced into a PCL/P123 electrospun substrate, and nanocomposites containing 4 wt% nano-filler were obtained. The corresponding properties of these polymer/ceramic nanocomposites, including morphology, thermodynamics, mechanics and biocompatibility, were evaluated. HA-F127/PCL/P123 showed superior mechanical performance, crystallinity percentage and thermal stability. The aforementioned improvements were primarily ascribed to a uniform dispersion of HA-F127 and strong interfacial adhesion between filler and matrix, which resulted from superior chain entanglements and interfacial crystallization of modified HA in the polymer substrate. Nanocomposites containing modified HA organized into a reliable platform for adhesion and proliferation of mouse L929 fibroblast cells. Finally, to analyze the interfacial interactions between phases, molecular dynamics simulation was applied. It was found that strong interfacial interactions exist between HA-F127 and PCL/P123. Consequently, HA-F127/PCL/P123 nanofibrous scaffolds can be considered as a promising candidate for tissue engineering applications.
1. Introduction
In the past few decades, there has been a growing need for bone regeneration because of various clinical bone diseases, such as bone infections, bone tumors and bone loss. Bone tissue engineering aims to renovate lost tissue by using a scaffold engineered to function as an extracellular matrix.1 Collagen protein fibers and nanosized calcium compounds constitute the bone tissue. Hence, significant attention has been paid to fabricate engineered scaffolds composed of biopolymers and nanophase hydroxyapatite (HA) because they can provide ideal conditions for cell growth and differentiation.2,3Hydroxyapatite (HA) is a type of calcium phosphate with the chemical composition Ca10(PO4)6(OH)2 which identically resembles that of natural bone mineral (Ca/P = 1.67). Due to HA's excellent properties, such as bioactivity, biocompatibility, osteoconductivity, nontoxicity, relatively stable degradation and mechanical strength, this bioceramic has found numerous applications as bone substitutes and tissue engineering scaffolds. In particular, HA not only plays a primary role in improving the mechanical properties of the nanocomposites, but also provides a favorable environment for osteoconduction, protein adhesion, and osteoblasts proliferation.4,5
In most cases due to the extremely high surface area in nanocomposites, an improvement of the mechanical and thermo mechanical properties, bioactivity and biocompatibility can be achieved at low filler contents which marks the main advantage of these materials over micro phase reinforced composites (less than 5 wt%).6,7 However, agglomeration and weak interfacial interactions of nano-reinforcements result in reduced stress transfer at the reinforcement-polymer interface and therefore the full potential of these materials cannot be fully exploited. In other words, nano-reinforcements and more especially nanoparticles with great specific surface tend to aggregate in polymer matrix, and the interface between organic polymer and inorganic nanoparticles is often weak because of the incompatibility. Simultaneously, incompatibility and phase separation of nanoparticles agglomerates result in morphology defects and failure at the interface and thus reduces nanocomposite biocompatibility.8,9
In polymer nanocomposites, nanoparticles surface functionalization has been proven to be a pertinent technique which minimizes particle/particle interaction and enhances particle/matrix interaction. Interfacial interaction has a more dominant role in case of nanocomposites, which determines the dispersion and agglomeration formation of the filler within the matrix and the stress transfer along the interface. This in turn dictates the physical properties of the polymer matrix and consequently the macroscopic characteristics of the nanocomposite. In order to improve the inorganic nanoparticles compatibility in polymeric substrates, surface modification with organic molecules seems to be relevant. Resultantly, the surface modified nanoparticles can disperse into polymeric matrix homogeneously. Moreover, the surface modification agent would interact with the matrix by chemical or physical interaction, thus the compatibility could be greatly improved.
Various methods have been utilized to tune the surface properties of HA, including the use of small molecules like silane coupling agents,10 dodecyl alcohol,11 etidronic acid,12 isocyanates,13 organophosphonic acids,14 carboxylic acids15 and polymer chains such as poly(hydroxyethyl methacrylate),16 poly(3-hydroxybutyrate),5 polyethylene glycol,17 poly(methyl methacrylate),18 polypyrrole,19 poly(acrylic acid),20 poly(N-isopropylacrylamide),21 collagen,22 polylactic acid23 and polycaprolactone.24 In these methods, the surface hydroxyl groups of HA directly and covalently bonded with the mentioned molecules and polymer chains following the relevant chemical reaction.
Polycaprolactone (PCL), a semi-crystalline linear polyester, has attracted growing interest for its biomedical applications in bone tissue engineering over the last few decades.25 Moreover pluronic, an ABA-type triblock copolymer consisting of hydrophilic polyethylene oxide (PEO) units and hydrophobic polypropylene oxide (PPO) units arranged in a linear triblock structure (PEO)x–(PPO)y–(PEO)x, has been used in approaches toward increasing blend hydrophilicity.26,27 Our previous study showed that PCL/P123 fibrous scaffolds not only improved hydrophilicity but also elongation at break and ultimate tensile strength of blended samples compared to the neat PCL scaffold. Therefore, the PCL/P123 is a promising polymer matrix for tissue engineering applications.28 The first aim of this study, therefore, was to covalently attach the pluronic F127 to the HA surface via esterification reaction. The structure obtained was studied using Fourier transform infrared (FTIR) spectroscopy, thermogravimetric analysis (TGA), X-ray diffraction (XRD) and transmission electron microscope (TEM). To the best of our knowledge, this is the first report on the attachment of pluronic F127 via covalent bonding to the surface of HA surface. The second aim was to embed neat and modified HA in the PCL/P123 solution and electrospun the resulted suspensions. Corresponding properties were evaluated, including morphology, mechanical and thermodynamics. Furthermore, to evaluate the influence of surface modification on the HA/polymer interactions at atomic level, molecular dynamic simulations were designed and evaluated.
2. Materials and methods
2.1. Materials
PCL (Mw = 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), pluronic F127 (Mw = 12600), pluronic P123 (Mn = 5600), hydroxyapatite nanocrystals with average diameter less than 200 nm (Mw = 502.31 g mol−1), maleic anhydride and manganese acetate were purchased from Sigma Aldrich. Diethyl ether, toluene, tetrahydrofuran, chloroform and methanol solvent were obtained from Merck. All reagents were of analytical grade and were used as received without any further purification.
000), pluronic F127 (Mw = 12600), pluronic P123 (Mn = 5600), hydroxyapatite nanocrystals with average diameter less than 200 nm (Mw = 502.31 g mol−1), maleic anhydride and manganese acetate were purchased from Sigma Aldrich. Diethyl ether, toluene, tetrahydrofuran, chloroform and methanol solvent were obtained from Merck. All reagents were of analytical grade and were used as received without any further purification.
2.2. Synthesis of F127-COOH
F127 (1.0 g) and maleic anhydride (0.17 g) were dissolved in distilled chloroform (7 mL) and the resulting solution was allowed to react for 24 h under stirring at 70 °C. Following completion of the reaction, the solution was concentrated and poured twice into an excess amount of ice cold diethyl ether to precipitate the reaction product. Then, this modified pluronic (F127-COOH) was dried and collected as white power.292.3. Immobilization of F127 on hydroxyapatite surface
The modification of hydroxyapatite (HA) surface was achieved by a condensation reaction with F127-COOH. A typical procedure for HA surface grafting is briefly described, as follows: firstly, 0.5 g HA was dispersed in tetrahydrofuran (3.75 mL), under ultrasonication at room temperature for 45 min. Then, 0.5 g F127-COOH in which was introduced 0.06 g manganese acetate as catalyst was slowly dropped into the above stirred suspension. The suspension was heated to 60–70 °C with reflux and maintained for 60 min, and then about 2 mL tetrahydrofuran was removed. Then 2 mL toluene were added into the reaction mixture, afterwards the mixture was heated and maintained at 120 °C for 18 h under constant stirring. After cooling at room temperature, the reaction mixture was washed several times with tetrahydrofuran to remove excess amount of ungrafted pluronic from hydroxyapatite particles, and finally the solid phase was isolated by centrifugation (8000 rpm) and filtered through 0.2 μm PTFE filter. The filtrate was dried under vacuum 24 h at 60–70 °C. The surface modified HA (HA-F127) powder was obtained.302.4. Electrospinning process
The polymer solution with concentration of 10 wt% was prepared by dissolving the mixtures of PCL and P123 in chloroform/methanol (3/1 in vol%) at 90/10 ratio to make PCL/P123 solutions. HA and HA-F127 was sonicated in 4% wt/wt PCL/P123 for 45 min. HA and HA-F127 were added to PCL/P123 solution, respectively to make HA/PCL/P123 and HA-F127/PCL/P123 solutions. These solutions were magnetically stirred at room temperature for 24 h to obtain a fair suspension. Scaffolds were produced by electrospinning; set-up consisted of a high voltage supply, a syringe pump and a collector. For scaffold fabrication, each sample of prepared solution and suspensions was placed in a syringe (5 mL) topped with a 22 gauge needle and then connected to 14 kV voltage. The mass flow rate was 1.5 mL h−1, and the distance between the tip and the collector was 12 cm. Electrospinning was carried out at room temperature (25 ± 2 °C). All electrospun fibers were deposited on a collector consisting of aluminum foil to form a thin nanofibrous membrane. The nanofibrous membranes were placed in vacuum drying at room temperature to completely remove any solvent residue.2.5. Cell culture method
The mouse L929 fibroblast cells were purchased from Pasteur Institute of Iran and used to study cell attachment and proliferation on scaffolds. The obtained cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and a 1% antibiotic (10000 units of penicillin) and incubated in humidified atmosphere of 5% CO2 at 37 °C.
2.6. In vitro cytotoxicity evaluation of nanocomposites
The specimens of PCL/P123, HA/PCL/P123 and HA-F127/PCL/P123 were cut into 1.5 cm diameter circular scaffolds, which were placed in 24-well tissue culture polystyrene (TCPS) and sterilized using autoclave. All of them were incubated at 37 °C in basal culture medium to prepare scaffolds (n = 3) in 3 days (1, 3 and 7 days). These collected extract scaffolds were replaced with culture medium of cells which had been seeded in 96-wells plate with initial density of 1 × 104 24 h ago. Then 10 μL MTT solutions were added to each well followed by incubation at 37 °C for 3 h. For dissolution of the dark-blue intracellular formazan, the supernatant was removed and 20 μL of DMSO (Merck) was added. The optical density was read spectrophotometrically at a wavelength of 492 nm. Mean of three measurements (n = 3) was selected to express the results and standard deviation was represented by error bars. A p value of less than 0.05 was considered statistically significant.2.7. Characterization
Fourier transform infrared (FTIR) spectra were recorded on a Bruker spectrophotometer within a range of 500–4400 cm−1 using a resolution of 4 cm−1. The samples were prepared on a KBr pellet in vacuum desiccators under a pressure of 0.01 Torr.Thermogravimetric analysis (TGA) was carried out using a Mettler Toledo TGA/SDTA851 under N2 atmosphere at a flow rate of 50 mL min−1. The thermograms were obtained from 0 to 700 °C at a heating rate of 10 °C min−1. A sample weight of about 10 mg was used for all the measurements.
X-ray diffraction (XRD) spectra were collected on an X-ray diffraction instrument (Siemens D5000) with a Cu target (k = 0.1540 nm) at room temperature. The system consists of a rotating anode generator, and operated at 35 kV and a current of 20 mA. The samples were scanned from 2 to 40° 2θ at the step scan mode, and the diffraction pattern was recorded using a scintillation counter detector. The basal spacing of the samples was calculated using the Bragg's equation.
The transmission electron microscope (TEM), Zeiss EM10C, with an accelerating voltage of 80 kV was employed to study the morphology of the neat and surface modified HA particles. Samples were prepared by sonication of HAs in ethanol and their deposition on a carbon coated copper grid (mesh 300).
The morphology of electrospun structures was determined by scanning electron microscopy (SEM) (SERON AIS-2100, South Korea) using 15 kV accelerating voltage. The samples were coated with gold using a sputter coater before SEM imaging. The average fiber diameter and corresponding size distribution were analyzed by measuring 200 fibers presented in SEM micrographs using an image analyzer (Image J, developed by the National Institute of Health, USA).
Differential scanning calorimetry (DSC) experiments were conducted with a Flash DSC 1 of Mettler-Toledo, in order to evaluate the thermal properties of the nanocomposites. The experiments were conducted at a constant heating rate of 10 °C min−1 on samples (15–20 mg) packed in aluminum pans under nitrogen flow. The samples were heated in two stages. First, the samples were heated to 120 °C and kept at that temperature for 5 min to erase the thermal history. Afterward they were cooled to −60 °C and then reheated again to 120 °C. The melting temperature (Tm) and the melting enthalpy (ΔH) of these nanocomposites were determined from the heating scans. The crystallinity percentage (% Xc) can be calculated by applying the equation:

 is the melting enthalpy of 100% crystalline PCL (139 J g−1) and w is the weight fraction of PCL in the nanocomposite.
is the melting enthalpy of 100% crystalline PCL (139 J g−1) and w is the weight fraction of PCL in the nanocomposite.
Mechanical properties of different scaffolds were performed using an electromechanical tensile tester (Instron 5566, Elancourt, France). Rectangular specimens with a length of 20 mm and a width of 5 mm were manually cut from electrospun samples. All samples were mounted between holders at a distance of 2 cm. The stress strain curves of these materials were constructed from the load-deformation curves recorded at a stretching speed of 5 mm min−1.
2.8. Simulation details
MD simulations were adjusted with the commercial molecular modeling software package Materials Studio 4.3 from Accelrys Software Inc. (San Diego, CA). For computing inter atomic interactions the COMPASS (condensed-phase optimized molecular potentials for atomistic simulation studies) force field was selected. It should be noted that, the COMPASS force field has been already applied to a wide range of materials such as PCL, pluronic and hydroxyapatite.31–33 PCL and P123 chains were built from their repeat units by Build module, and then cubic simulation boxes were constructed using Amorphous cell module. To obtain a global energy minimized state, the PCL/P123 structure was annealed by performing dynamics simulations for 500 ps at each temperature increased from 298 K to 500 K and then decreased to 298 K again with a step of 40.4 K. Finally, the annealed PCL and P123 chains were used in the following interaction study. In order to build the crystal structure of HA on the MD platform, the space group was set to P63/m with the lattice parameters as a = b = 9.424 Å, c = 6.879 Å. In the HA crystal, the OH ions were formed into arrays with the direction parallel to the c-axis; each OH ion was coordinated to the three Ca atoms, which composed a triangle on a same plane. Viewed along the c-axis, each two adjacent Ca atoms could be matched together after 60° rotation, located on two different planes. The cell constants calculated for HA crystal was in good agreement with the experimental values (data are not shown).34 The crystallographic surface, (1 1 0), was chosen to study the interaction of modified and pristine simulated HA nanoparticles with PCL/P123 substrate. The HA surface was built by cleaving the crystal along the crystallographic plane and then minimized under periodic boundary conditions. The chemical functionalization of the HA was performed by attaching pluronic F127-COOH chains to the surface through chemical covalent bonding. Grafted F127 chains were attached to the HA surface oxygen atoms and H2O molecules were removed. Since functionalized HA was in a relatively high-energy state, minimizations was performed to remove unwanted interactions and to attain the lowest energy state. For this purpose, functionalized HA was optimized by the steepest descent and conjugate gradient method. To create the entire system, a simulation box was generated and both the optimized PCL/P123 chains and the functionalized HA plane were included in it; the functionalized HA plane was put at the bottom of the simulation box for visual convenience. A vacuum space with the height of 100 Å was added on top of the PCL/P123 chains to restrict its interaction to only the uppermost atom layer of functionalized HA and also to enhance computational efficiency. Again, to omit the undesirable interactions, including overlapping and close contact, a 20000 step energy minimization was adopted using the steepest descent and conjugate gradient method. In addition, all of the atoms in the polymer bulk and HA surface were kept constrained by fixing the Cartesian coordinates throughout the minimization and dynamics steps. To calculate interaction of the HA with the polymer chains, MD simulation was performed using an NVT ensemble for 2000 ps, where the temperature was controlled by the Anderson thermostat (298 K). Discover module was used for minimization and dynamic parts. The integration of the equations of motion has been performed by means of the Verlet velocity time integration method with a time step of 1 femtosecond (fs). The initial velocities of the atoms were assigned using a Maxwell–Boltzmann distribution at the desired temperature and pressure. A cutoff radius of 1.25 nm was used for Lennard-Jones interactions and Ewald summation to compute long-range electrostatic interactions.
3. Results and discussion
3.1. Characterization of HA and HA-F127 nanoparticles
In order to decrease the agglomerates of hydroxyapatite nanoparticles and improve their distribution and dispersion in polymeric matrix, surface grafting of HA nanoparticles with F127-COOH and its further incorporation in PCL/P123 fibrous scaffold were carried out in this study. The first step of this process was to modify the F127. Briefly, the intermediate product F127-COOH was first synthesized through converting the terminal hydroxyl groups of the F127 chains into carboxyl groups by reacting with maleic anhydride. Then, from the reaction of carboxyl groups of F127-COOH with the hydroxyl groups of hydroxyapatite surface, ester linkages were obtained. Fig. 1 schematically illustrates a possible reaction mechanism for HA modification. | ||
| Fig. 1 Reaction routes for surface modification of HA nanoparticle (a) synthesis of F127-COOH, (b) immobilization of F127 on hydroxyapatite surface. | ||
Fig. 2a shows the FTIR spectra of F127 and F127-COOH. As seen, a sharp peak appears at 1730 cm−1 in F127-COOH spectra which can be attributed to the newly formed carbonyl group in this sample. Therefore, successful transformation of F127 hydroxyl groups to carboxyl groups through interaction with maleic anhydride can be postulated.
 | ||
| Fig. 2 FTIR spectra of (a) F127 and F127-COOH and (b) HA and HA-F127 samples. | ||
FTIR spectra of HA and HA-F127 are presented in Fig. 2b. Considering HA spectrum, the absorption peaks at 562 and 603 cm−1 are assigned to the deformation vibrations of phosphate groups whereas peaks at 1028 and 962 cm−1 are attributed to stretching vibrations of these groups. The band at 3565 cm−1 is related to the stretching vibration of free hydroxyl group. The small adsorption peak around 1600 to 1650 cm−1 and the broad adsorption band at 3412 cm−1 are assigned to the bending vibrations and stretching vibrations of surface absorbed and structural H2O in the HA, respectively.30 From Fig. 2b, a new adsorption peak at 1730 cm−1 appears which can be assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of grafted F127-COOH. Moreover, peaks in the range of 3000–2850 cm−1 are corresponded to the C–H stretching vibrations of the CH3 and CH2 groups of grafted polymer chains. Since HA nanoparticles were repeatedly washed with tetrahydrofuran after the modification process, the existence of ungrafted F127-COOH remanence in the HA-F127 sample can be ignored and therefore the newly observed peaks in HA-F127 spectrum can be solely attributed to the surface grafted polymer chains.
O stretching vibration of grafted F127-COOH. Moreover, peaks in the range of 3000–2850 cm−1 are corresponded to the C–H stretching vibrations of the CH3 and CH2 groups of grafted polymer chains. Since HA nanoparticles were repeatedly washed with tetrahydrofuran after the modification process, the existence of ungrafted F127-COOH remanence in the HA-F127 sample can be ignored and therefore the newly observed peaks in HA-F127 spectrum can be solely attributed to the surface grafted polymer chains.
TGA analysis was used to study the thermal properties of the HA samples and also the related modifier content. The amount of surface grafted polymer was determined as a weight loss percentage during heating. TGA and dTG results for the pristine F127, neat and modified HA are shown in Fig. 3. According to the results, a small mass loss of HA samples at temperatures less than 100 °C can be attributed to the absorbed water molecules on the surface of nanoparticles. This mass loss is continuous for HA until 520 °C whereas no mass loss can be observed for the modified HA in the range of 100 °C to 220 °C suggesting the omission of hydroxyapatite structural water molecules during modification process. The second stage of neat HA mass loss is observed in the temperature range of 520 to 700 °C, which results from dehydroxylation of surface and structural OH groups. The beginning of this stage shifts to the temperature of 580 °C for the modified HA sample suggesting improved thermal stability of modified HA nanoparticles. The considerable mass loss in the range of 220 to 580 °C for HA-F127 sample is due to the thermal decomposition of the grafted F127. The neat and modified HA sample reaches to 97.51% and 93.84% char value at 700 °C. Thermal decomposition of F127 is observed gradually in the temperatures range 220–580 °C. Also, weight loss at the dehydroxylation step is lower for HA-F127, which results from the covalent reaction of hydroxyl groups with F127. Moreover, pristine F127 completely pyrolyzes before the beginning of surface and structural OH groups of hydroxyapatite dehydroxylation. Compared to pristine F127 degradation temperature, the mass loss of attached F127 chains in HA-F127/PCL/P123 TGA curve obviously shifts to higher temperature and the thermal degradation temperature remarkably increases from 245 to 300 °C. This may be due to the decrease in mobility of F127 chain grafted to hydroxyapatite nanoparticles. The amount of F127 grafted on the HA nanoparticle surfaces was about 4 wt%.
 | ||
| Fig. 3 TGA and dTG curves of pristine F127 (offset), neat and functionalized HA nanoparticles. | ||
The surface modification of HA nanoparticle should occur only at its surface, in order to keep the state of the bulk properties such as crystallinity and the crystalline phase, intact. If the chemical reaction alters the state of the bulk properties the resulting HA nanoparticle loses its intrinsic properties. Thus, confirmation of the selective reaction on the HA nanoparticle surface and the maintenance of crystalline properties are important. XRD was used to characterize the crystallinity of HA and HA-F127 samples. As shown in Fig. 4, the characteristic diffraction peaks of the (002), (211), (300), (202), (310), (222), (213), and (411) crystal planes of HA nanoparticle is not different from that of HA-F127. This observation demonstrated that grafting reaction did not alter the crystalline structure of HA nanoparticles. However, intensity of the mentioned peaks decreases by a considerable value as a result of polymer grafting. Similar observation has been made in Hemmatpour et al. study.35 They stated that grafting poly (N,N-dimethyl amino ethyl methacrylate) on halloysite nanotube surface diminishes the intensity of the halloysite characteristic peaks. Halloysite, a naturally occurring polymorph of kaolinite, have hydroxyl as its surface functional groups similar to hydroxyapatite.
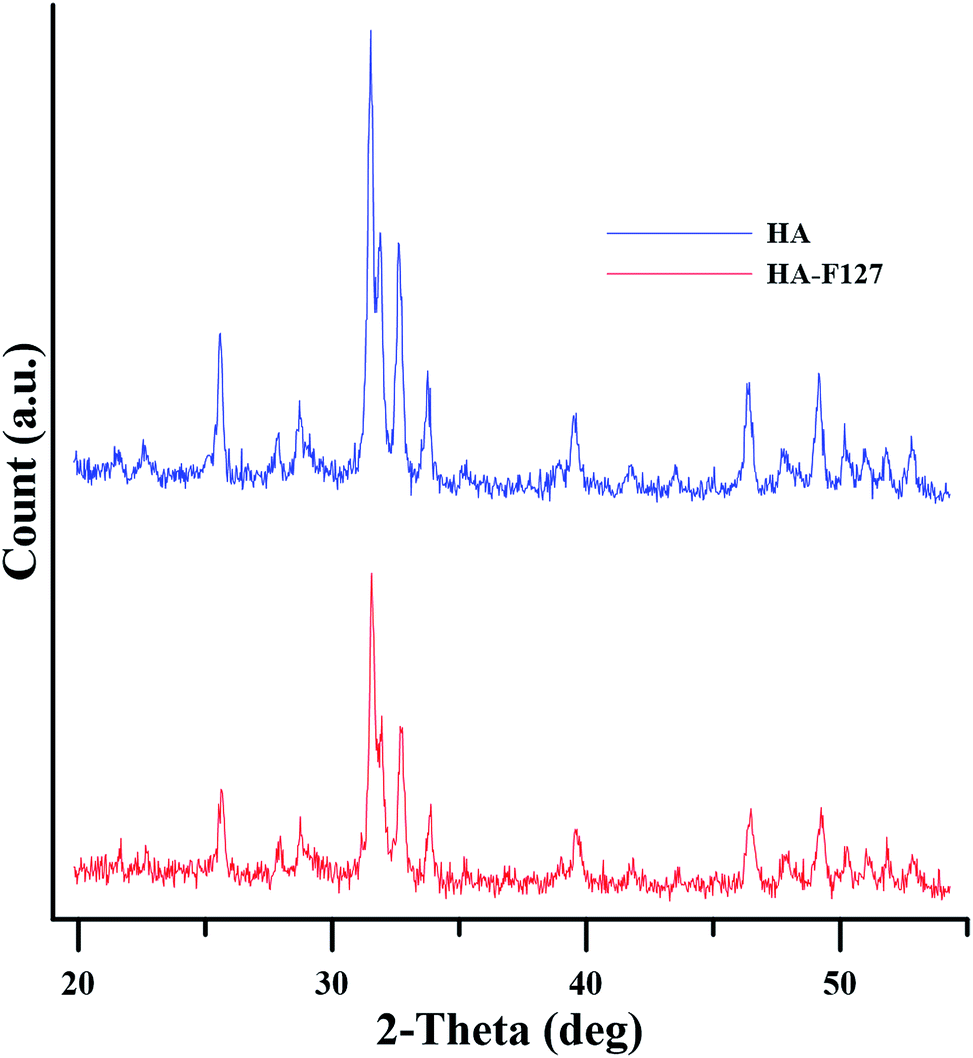 | ||
| Fig. 4 XRD patterns of neat and modified HA nanoparticles. | ||
Transmission electron microscopy (TEM) was employed to study the morphology, size and dispersion of nanoparticles. Fig. 5 demonstrates that HA-F127 have spherical morphology with diameter of about 60 nm. Keen observation on the related TEM images of modified HA nanoparticles reveals once again a spherical morphology plus a 13 nm thick F127-COOH layer with smooth appearances, comprising a well formed core shell structure. This corona is more transparent than the HA nanoparticle, resulted from easier electron transfer, suggesting that shell composition is organic. Therefore successful surface modification of HA nanoparticles with F127-COOH chains can be concluded from TEM images.
 | ||
| Fig. 5 TEM image of HA-F127. | ||
The TEM images of HA and HA-F127 nanoparticles dispersed in ethanol with smaller magnification are shown in Fig. 6. The agglomerates of HA nanoparticles are evident from Fig. 6a while finely dispersed HA-F127 nanoparticles constructs the morphology presented in Fig. 6b. This can be attributed to the immobilization of F127 chains on HA surface, which improves their distribution and dispersion in solvent media by reducing the particle/particle interactions.
 | ||
| Fig. 6 TEM images of (a) HA and (b) HA-F127. | ||
Investigation on colloidal stability of HA samples is important because HA nanoparticles tend to aggregate due to van der Waals interactions as well as hydrogen bonding of surface hydroxyl groups. These possibilities can result in rapid precipitation of HA nanoparticles after distribution in the solvent media.36 Furthermore, to achieve a homogenous nanocomposite, high colloidal stability is crucial during the electrospinning of HA/polymer suspension. Monitoring of time-dependent colloidal stability of HA and HA-F127 in chloroform is illustrated in Fig. 7. After being stirred for 4 h in chloroform at room temperature, all HA and HA-F127 dispersed and suspended in the solvent. However, the aggregation and precipitation of HA nanoparticles started immediately after the stirring process halted. In the case of HA-F127 nanoparticles, the suspension remained fairly stable up to several hours, due to the non-polar interactions between the stretched F127 brushes and chloroform molecules. It is noteworthy to mention that F127 solubilizes in chloroform rather than forming stable micelles. In fact, the amphiphilic characteristic of the HA-F127 shell, inherited from F127, can cause colloidal stability in both aqueous and non-aqueous media. The existence of a polymer layer on the surface of modified HA nanoparticles overwhelms the overall HA density and inter-particle interactions, which favors the stability of nanoparticles in the solvent media. This argument is in good agreement with the dispersion state of nanoparticles in the TEM images.
 | ||
| Fig. 7 Monitoring of time-dependent colloidal stability of HA and HA-F127: (a) 5, (b) 30, (c) 60 minutes, and (d) 12 hours. | ||
3.2. Characterization of electrospun scaffolds
SEM images of PCL/P123, HA/PCL/P123 and HA-F127/PCL/P123 are presented in Fig. 8. As seen, scaffold of PCL/P123 demonstrates fibers in the range of 500 to 800 nm with smooth cylindrical morphology and no evidence of beads on fibers. The unimodal fiber diameter distribution of PCL/P123 scaffold is a direct effect of P123 incorporation into PCL fibers. P123 which has much lower molecular weight in comparison with that of PCL drastically diminishes the viscoelastic forces of the electrospinning solution and as a result surface tension role in determining the morphology of the resulted fibers becomes more significant.28 After HA incorporation into PCL/P123 electrospinning solution, the related average fiber diameter slightly increases. Moreover, due to the poor distribution and dispersion of neat HA in the electrospinning solution, agglomerates of HA nanoparticles are observable along the fibers and appear in short intervals of distance. The existence of nanoparticles agglomerates in fibers is an inherent characteristic of HA/polymer suspension electrospinning.37 It can be postulated that HA nanoparticles did not affect the electrical conductivity of the suspension; therefore the dominant morphology does not differ from that of PCL/P123. Surface modification decreases the agglomerates of HA nanoparticles and improves their dispersion in the electrospinning polymer solution. Therefore HA-F127/PCL/P123 morphology display finely dispersed HA-F127 along the electrospun fibers with no evidence of nanoparticles agglomeration. Moreover improved phase blending is expected between the modified HA shell (F127) and the P123 exist in electrospinning solution due to polymer chain similarity. | ||
| Fig. 8 SEM images of neat PCL/P123 fibers and nanocomposites of HA/PCL/P123 and HA-F127/PCL/P123. The fine dispersion of modified HA nanoparticles in PCL/P123 electrospun fibers and the absence of agglomerates are evident in HA-F127/PCL/P123 scaffold. | ||
The differential scanning calorimeter (DSC) test was performed to determine the thermal properties of electrospun nanocomposites. DSC experiments included a heating scan that gives information about the crystalline structure with electrospinning process history. This scan was performed between −60 °C and 120 °C. This was followed by a cooling procedure before the finale second heating scan which was recorded to characterize the structure of the polymer crystallized from the melt without the electrospinning process history. The thermal behavior of nanocomposites was characterized and the data were summarized in Table 1.
| Samples | First heat scan | Second heat scan | δ | ||||
|---|---|---|---|---|---|---|---|
| ΔH′ (J g−1) | % Xc | Tm (°C) | ΔH′′ (J g−1) | % Xc | Tm (°C) | ||
| PCL/P123 | 53 | 42.37 | 58.13 | 40 | 31.98 | 53.92 | 13 |
| HA/PCL/P123 | 60 | 47.97 | 58.14 | 42 | 33.58 | 54.00 | 18 |
| HA-F127/PCL/P123 | 77 | 61.55 | 59.63 | 55 | 43.97 | 55.01 | 22 |
During the first heating scan the crystallinity of PCL is affected by three parameters including the electrospinning process, imparting induced crystallization to PCL chains placed on the surface of developing jet, existence of HA nanoparticles in the vicinity of evolving PCL crystals and finally the HA surface modification. The latter has a drastic effect on the PCL nucleation and confined crystallization. The area under the DSC thermograms related to the PCL melting transition for different electrospun samples has been calculated. The difference between the mentioned area during first and second heat scans was obtained by the following equation described elsewhere:28
| δ = ΔH′ − ΔH′′ |
Parameter δ considerably increases for samples containing HA nanoparticles. This parameter signifies the electrospinning process effect on the formation of PCL crystals. The mentioned increment of parameter δ for HA/PCL/P123 and HA-F127/PCL/P123 samples in comparison with the PCL/P123 sample should be investigated through the effect of the presence of HA nanoparticles on the electrospinning solution properties. Inertness of HA nanoparticles dismisses the idea of these particles can be affected in the electrical field during electrospinning process, forming an aligned arrangement and consequently changing the amount of PCL crystallinity. However, incorporation of nanoparticle into electrospinning solution has a drastic effect on the overall viscosity.39 Simultaneously, due to the direct relationship between solution viscosity and shear rate, it can be postulated that presence of nanoparticles in the electrospinning solution enhances polymer chain alignment and induced crystallization. This can be the reason behind the observed increase in parameter δ by introducing HA nanoparticles into solution. Moreover, increased solution viscosity is expected when using modified nanoparticles which can establish strong polymer/particle and solvent/particle interfacial interaction. Therefore, by the same logic, incorporation of HA-F127 should have a more pronounced effect on solution viscosity, the resulted shear rate and finally induced crystallization of PCL chains in comparison with neat HA; justifying the observed 4 unit increase of parameter δ (see Table 1).
TGA and dTG curves of PCL/P123, HA/PCL/P123 and HA-F127/PCL/P123 were obtained (ESI Fig. 1†). The related weight loss of P123 which normally occurs in the range of 300 to 350 °C is merged with that of PCL which happens in the range of 350 to 450 °C. This may be due to the excellent compatibility and miscibility of these two polymers which approximate their thermal stability. In addition, synergistic interactions between PCL and P123 chains at 90/10 blend ratio have been reported previously and can also explain the mass loss behavior of PCL/P123.28 Blend of PCL and P123 fully pyrolyzes at the final temperature (700 °C). As a result, the final residual weight of nanocomposites corresponds to the neat and modified hydroxyapatite weight in their related TGA curves. The percentage weight of hydroxyapatite nanoparticles embedded in the HA-F127/PCL/P123 differs slightly from that of HA/PCL/P123 (0.5 wt%). Considering the results from colloidal stability experiment, this observation was anticipated as neat hydroxyapatite nanoparticles tend to precipitate in the polymer solution during the electrospinning process. Therefore, the initial concentration of neat hydroxyapatite nanoparticles used to fabricate the HA/PCL/P123 changes in the final scaffold. Surprisingly, the percentage weight of modified hydroxyapatite nanoparticles implanted in HA-F127/PCL/P123 suggests that most of the initially added HA-F127 nanoparticles enter the PCL/P123 fibers.
Scaffold manageable handling and load endurance are crucial during cell culture and therefore mechanical properties of the nanofibrous membrane play an important role on its successful application in tissue engineering. In most bone tissue engineering approaches, improving mechanical properties by incorporation of nanoscale reinforcements into substrates has been proven to be most effective. At high filler contents agglomeration of nanoparticle is inevitable due to the strong interactions between the nanoparticles themselves and therefore low filler content must be beneficial to the satisfactory dispersion of the nanoparticles for both untreated and grafted HA. The mechanical properties of nanocomposites mostly depend on the characteristics of components, composition, structure and interfacial interactions between the constituents.
Based on the typical stress–strain curves of the electrospun fibrous scaffolds (Fig. 9), tensile strength, elongation at break, and Young's modulus could be calculated (Table 2). Electrospun fibrous scaffolds containing nanoparticles show higher Young's modulus whereas their elongation at break is less than that of PCL/P123 sample. It is noteworthy to mention that adding HA nanoparticles to PCL/P123 results in decrease in tensile strength, implying that HA/PCL/P123 is more brittle than PCL/P123. This can be explained by heterogeneous distribution of HA nanoparticles, agglomerates and weak particle/matrix interfacial adhesion. On the contrary, for the samples containing the same amount of reinforcing phase, HA-F127/PCL/P123 shows higher tensile strength and Young's modulus which confirms the remarkable role of grafted F127 chains in enhancing mechanical properties of nanocomposite by reducing the particle–particle interactions and increasing particle–matrix interactions. This leads to uniform dispersion of nanoparticles, enhanced interfacial crystallization and consequently improved interfacial adhesion. Interfacial interactions can be described in a more quantitative way using the experimentally determined tensile properties. The theoretical model proposed by Pukánszky40,41 gave a quantitative relation between tensile strength and interfacial interaction as

 | ||
| Fig. 9 Stress–strain curves of fibrous scaffolds. | ||
| Samples | Tensile strength (MPa) | Young's modulus (MPa) | Elongation at break (%) | Bσy |
|---|---|---|---|---|
| PCL/P123 | 3.6 ± 0.2 | 4.6 ± 0.4 | 163.4 ± 6.7 | — |
| HA/PCL/P123 | 3.0 ± 0.2 | 6.6 ± 0.1 | 88.1 ± 5.3 | −1.15 ± 0.15 |
| HA-F127/PCL/P123 | 5.0 ± 0.3 | 10.2 ± 0.2 | 103.8 ± 4.4 | 11.61 ± 0.11 |
3.3. Cell adhesion and proliferation studies
MTT assay was performed to investigate the biocompatibility of collected extract nanocomposites. Fig. 10 illustrates cell densities on the surfaces of PCL/P123, HA/PCL/P123 and HA-F127/PCL/P123 as a function of the cell culture time. In the first day, all of the scaffolds showed similar cell attachment behavior. However, cell count on HA/PCL/P123 showed a slight decrease in comparison to PCL/P123 in the next stage. This observation suggests that the incorporation of neat HA nanoparticles in the polymer nanofibrous scaffold can negatively affect the cell attachment, which may be initiated by the nanoparticle agglomeration and the subsequent defect formation in scaffold morphology, especially at the initial stages of the cell culture study.42 In general, utilizing the HA-F127 relatively contributed to the cell proliferation, but not effectively up to three days. As the incubation time progresses, cell proliferation on samples containing nanoparticles becomes more pronounced than PCL/P123. Lucidly, sample comprising modified HA provides better surfaces for cell growth than that of other samples as the incubation time increases. The enhanced interfacial adhesion of modified HA with PCL/P123 substrates is the underlying cause of the mentioned observation. Moreover, permeation of the culture media into fibers and fiber swelling during cell culture studies ease the aggregation of neat HA nanoparticles and consequently exclusion of nanoparticles from polymer substrate take form. Indeed when incubation time increases, HA nanoparticles, whether being modified or neat, enhance the cell attachment and proliferation significantly because of their excellent bioactive and biocompatible nature. Furthermore, increased fiber surface roughness is expected when using nanoparticles; therefore in the last day of incubation, scaffolds containing neat and modified HA shows substantial cell density.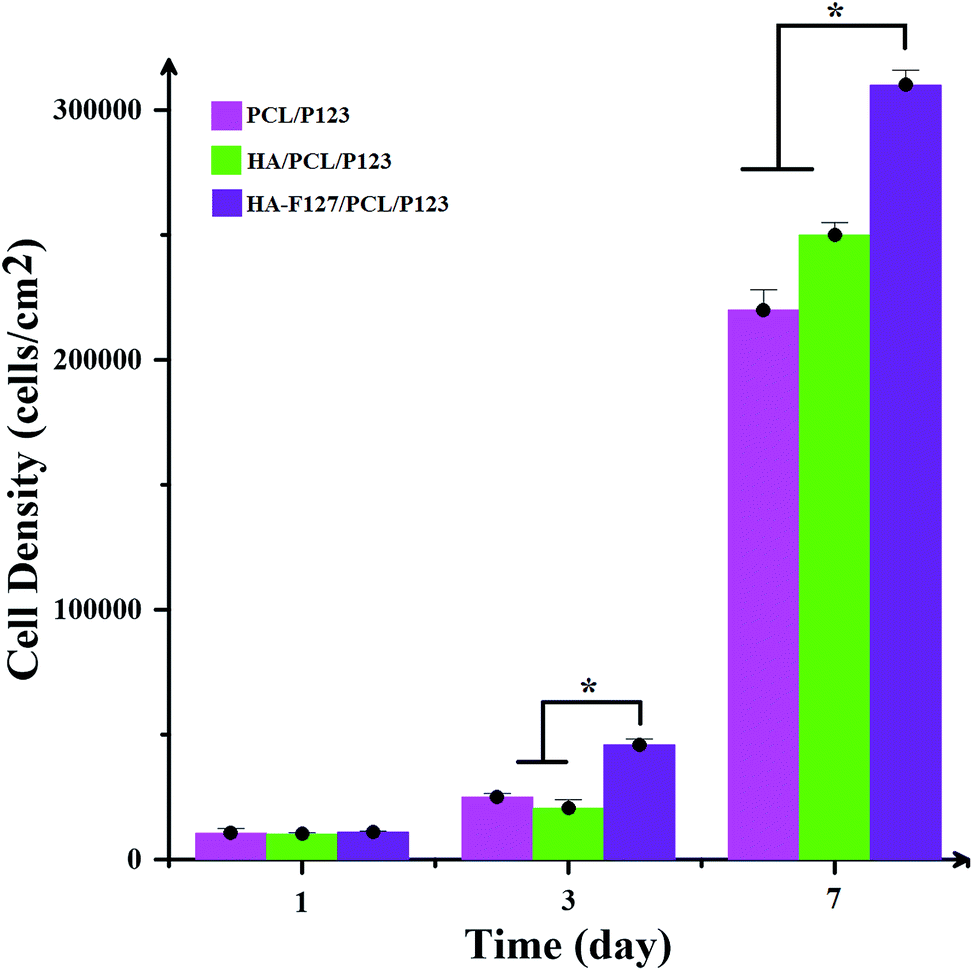 | ||
| Fig. 10 Cell adhesion and proliferation on various classes of substrates: asterisk shows significant difference with p < 0.05. | ||
3.4. MD simulation
The interfacial characteristics between the nanofiller and polymer substrate have tremendous effects on the nanocomposite properties. Hence, the structural properties of the polymer in the vicinity of a nanoparticle have been studied. To gain a better understanding of the interfacial interaction, the MD simulation was applied (Fig. 11). The interaction energy between the HA nanoparticle and PCL/P123 substrate at the end of the equilibration process can be obtained:| Einteraction = Enanocomposite − Ematrix − Enanoparticle |
 | ||
| Fig. 11 The hierarchical order of MD simulation for HA/PCL/P123 and HA-F127/PCL/P123 systems. | ||
The interaction energy of the nanoparticle with the polymer chains in each system was calculated, and results were displayed in Table 3. Interactions of PCL/P123 chains with HA-F127 are about 1.5 times larger than that of the pure HA (absolute value). By comparing these values and since more negative interaction energy indicates larger attraction, it can be concluded that the interaction between HA-F127 and PCL/P123 chain is stronger than that of pristine HA. Elevated interaction energy of HA and polymer substrate after the modification process promises better distribution and dispersion of nanoparticles in fibrous scaffold. This speculation was confirmed by the results from the colloidal stability of the modified HA nanoparticles and the interfacial interaction parameter calculated from Pukanszky model and are in line with the MD simulations data.
| System | Enanocomposite | Ematrix | Enanoparticle | Einteraction |
|---|---|---|---|---|
| HA/PCL/P123 | 1539807.307168 |
7755.181379 | 1569923.851001 |
−37871.725212 |
| HA-F127/PCL/P123 | 1556734.141610 |
13438.564580 |
1599935.254224 |
−56639.677194 |
4. Conclusion
In order to enhance the interaction and subsequent distribution and dispersion of HA nanoparticles in the polymer substrate, HA-F127 with a core–shell structure was successfully synthesized by immobilizing F127-COOH onto hydroxyapatite surface. The grafting reaction was verified by FTIR, TGA and TEM analysis. Modified HA FTIR spectra showed some new peaks which were appeared at 1730 cm−1 and in the range of 3000–2850 cm−1, corresponding to CO stretching vibration and C–H stretching vibrations of the CH3 and CH2 groups of grafted polymer chains, respectively. The grafting ratio of immobilized F127 was 4 wt%. XRD analysis demonstrated that grafting reaction did not alter the crystalline structure of HA nanoparticles and surface modification of HA nanoparticle occurred only at its surface. Nanocomposites comprising HA and HA-F127 were prepared by electrospinning. Although a same amount of pristine and modified nanoparticles was used to fabricate scaffolds, PCL crystallinity percentage has increased from 47.97% in HA/PCL/P123 to 61.55% in HA-F127/PCL/P123. Compared to HA/PCL/P123, the modified nanocomposites tensile strength and Young's modulus were improved by 67% and 55%, respectively. At the same time, the Tm increased by about 2 °C. These observations confirmed the remarkable role of grafted F127 chains in enhancing interfacial adhesion between polymer matrix and nanofiller. MD simulation revealed that HA-F127 has more dominant interfacial adhesion with the PCL/P123 matrix than the neat HA due to higher interaction energy. In vitro biological evaluation showed that the existence of modified HA in nanocomposites favors cell adhesion and proliferation. These results suggest that HA-F127/PCL/P123 scaffold could be considered as an ideal candidate for bone tissue engineering.
References
- C. Chaput, A. Selmani and C. H. Rivard, Curr. Opin. Orthop., 1996, 7, 62–68 CrossRef.
- A. Abdal-hay, H. R. Pant and J. K. Lim, Eur. Polym. J., 2013, 49, 1314–1321 CrossRef CAS.
- K. Rezwan, Q. Chen, J. Blaker and A. R. Boccaccini, Biomaterials, 2006, 27, 3413–3431 CrossRef CAS PubMed.
- M. Okada and T. Furuzono, Sci. Technol. Adv. Mater., 2012, 13, 064103 CrossRef.
- M. Szubert, K. Adamska, M. Szybowicz, T. Jesionowski, T. Buchwald and A. Voelkel, Mater. Sci. Eng. C, 2014, 34, 236–244 CrossRef CAS PubMed.
- J. Hui and X. Wang, Inorg. Chem. Front., 2014, 1, 215–225 RSC.
- T. Kuilla, S. Bhadra, D. Yao, N. H. Kim, S. Bose and J. H. Lee, Prog. Polym. Sci., 2010, 35, 1350–1375 CrossRef CAS.
- M. Rong, M. Zhang and W. Ruan, Mater. Sci. Technol., 2006, 22, 787–796 CrossRef CAS.
- S. Kango, S. Kalia, A. Celli, J. Njuguna, Y. Habibi and R. Kumar, Prog. Polym. Sci., 2013, 38, 1232–1261 CrossRef CAS.
- S. Wang, S. Wen, M. Shen, R. Guo, X. Cao, J. Wang and X. Shi, Int. J. Nanomed., 2011, 6, 3449–3459 CAS.
- L. Borum-Nicholas and O. Wilson, Biomaterials, 2003, 24, 3671–3679 CrossRef CAS PubMed.
- M. Othmani, A. Aissa, C. G. Bac, F. Rachdi and M. Debbabi, Appl. Surf. Sci., 2013, 274, 151–157 CrossRef CAS.
- Q. Liu, Hydroxyapatite/polymer composites for bone replacement, University of Twente, 1997 Search PubMed.
- S. C. D'Andre and A. Y. Fadeev, Langmuir, 2003, 19, 7904–7910 CrossRef.
- L. Chen, J. M. Mccrate, J. C. Lee and H. Li, Nanotechnology, 2011, 22, 105708 CrossRef PubMed.
- K. Adamska, M. Szubert, A. Voelkel and Z. Okulus, Chem. Pap., 2013, 67, 429–436 CAS.
- Q. Liu, J. R. de Wijn, K. de Groot and C. A. van Blitterswijk, Biomaterials, 1998, 19, 1067–1072 CrossRef CAS PubMed.
- Y. Wang, X. Zhang, J. Yan, Y. Xiao and M. Lang, Appl. Surf. Sci., 2011, 257, 6233–6238 CrossRef CAS.
- S. Yala, H. Khireddine, D. Sidane, S. Ziani and F. Bir, J. Mater. Sci., 2013, 48, 7215–7223 CrossRef CAS.
- C. S. Goonasekera, K. S. Jack, J. J. Cooper-White and L. Grøndahl, J. Mater. Chem. B, 2013, 1, 5842–5852 RSC.
- J. Wei, P. He, A. Liu, X. Chen, X. Wang and X. Jing, Macromol. Biosci., 2009, 9, 1237–1246 CrossRef CAS PubMed.
- Z.-C. Xing, K.-W. Chang, S. Chun, S. Kim and I.-K. Kang, Tissue Eng. Regener. Med., 2014, 11, 99–105 CrossRef CAS.
- Y. Hu, X. Gu, Y. Yang, J. Huang, M. Hu, W. Chen, Z. Tong and C. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 17166–17175 CAS.
- L. Zeng, H. Wang, G. Fu, J. Jiang and X. Zhang, J. Colloid Interface Sci., 2010, 352, 36–42 CrossRef CAS PubMed.
- M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC.
- S. S. Zargarian and V. Haddadi-Asl, Artif. Cells, Nanomed., Biotechnol., 2016, 1–10 CrossRef PubMed.
- S. S. Zargarian and V. Haddadi-Asl, Int. J. Polym. Anal. Charact., 2016 DOI:10.1080/1023666X.2016.1192837.
- M. Mirhosseini, V. Haddadi-Asl and S. S. Zargarian, J. Appl. Polym. Sci., 2016, 133(17) DOI:10.1002/app.43345.
- Y.-Y. Li, L. Li, H.-Q. Dong, X.-J. Cai and T.-B. Ren, Mater. Sci. Eng. C, 2013, 33, 2698–2707 CrossRef CAS PubMed.
- N. Tudorachi and A. P. Chiriac, J. Polym. Environ., 2011, 19, 546–558 CrossRef CAS.
- C. Gu, H. Gu and M. Lang, Macromol. Theory Simul., 2013, 22, 377–384 CrossRef CAS.
- S. Dong, X. Cui, S. Zhong, Y. Gao and H. Wang, Mol. Simul., 2011, 37, 1014–1022 CrossRef CAS.
- H.-p. Zhang, X. Lu, Y. Leng, L. Fang, S. Qu, B. Feng, J. Weng and J. Wang, Acta Biomater., 2009, 5, 1169–1181 CrossRef CAS PubMed.
- J. M. Hughes, M. Cameron and K. D. Crowley, Am. Mineral., 1989, 74, 870–876 CAS.
- H. Hemmatpour, V. Haddadi-Asl and H. Roghani-Mamaqani, Polymer, 2015, 65, 143–153 CrossRef CAS.
- H. J. Lee, H. W. Choi, K. J. Kim and S. C. Lee, Chem. Mater., 2006, 18, 5111–5118 CrossRef CAS.
- S. S. Zargarian and V. Haddadi-Asl, Iran. Polym. J., 2010, 19, 457–468 CAS.
- N. Ning, S. Fu, W. Zhang, F. Chen, K. Wang, H. Deng, Q. Zhang and Q. Fu, Prog. Polym. Sci., 2012, 37, 1425–1455 CrossRef CAS.
- S.-P. Sun, M. Wei, J. R. Olson and M. T. Shaw, Rheol. Acta, 2011, 50, 65–74 CrossRef CAS.
- X.-L. Xie, C.-Y. Tang, X.-P. Zhou, R. K.-Y. Li, Z.-Z. Yu, Q.-X. Zhang and Y.-W. Mai, Chem. Mater., 2004, 16, 133–138 CrossRef CAS.
- B. Pukánszky, B. Turcsanyi and F. Tudos, Interfaces in polymer, ceramic and metal matrix composites, Elsevier, Amsterdam, 1988, pp. 467–477 Search PubMed.
- Z. Hong, P. Zhang, C. He, X. Qiu, A. Liu, L. Chen, X. Chen and X. Jing, Biomaterials, 2005, 26, 6296–6304 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra19499k |
| This journal is © The Royal Society of Chemistry 2016 |