
Greatly enhanced photocatalytic activity of semiconductor CeO2 by integrating with upconversion nanocrystals and graphene
Abstract
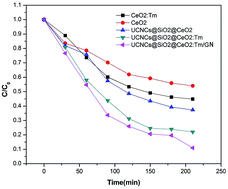
CeO2 is an important semiconductor photocatalyst that has been extensively utilized in photocatalysis. However, its photocatalytic efficiency is still not desirable for practical applications with solar light due to the low light absorption efficiency and high recombination rate of photogenerated electron–hole pairs. Up to now, many approaches have been developed to improve the photocatalytic activity of CeO2 and much progress has been made. However, the NIR light that accounts for 46% of the total solar light is unutilized for photocatalysis. In the present study, greatly enhanced photocatalytic activity of CeO2 by integrating upconversion nanocrystals and graphene into CeO2 was achieved for the first time. A novel nanocomposite photocatalyst of NaLuF4:Gd,Yb,Tm@SiO2@CeO2:Tm/graphene was developed, which took advantage of a synergetic effect to enhance the photocatalytic activity of CeO2. The upconversion nanocrystals (UCNCs) absorb the NIR light and transfer energy to CeO2, thus extending the light-responsive range of CeO2 to the NIR region and making CeO2 produce highly energetic electron–hole pairs. Tm-doping narrows the band-gap of CeO2, resulting in a red-shift of the absorption edge for CeO2. Graphene enhances adsorption of pollutants and serves as an effective electron acceptor and transporter. This work provides new insights into the greatest improvement of catalytic activity of CeO2 through effective integration of upconversion material, CeO2, and graphene into a hetero-nanocomposite structure. This strategy can be widely used to fabricate semiconductor-based nanocomposite photocatalysts with high photocatalytic activities and facilitate their applications in environmental protection issues using solar light.
 Please wait while we load your content...
Please wait while we load your content...

