
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe first solvent-free synthesis of privileged γ- and δ-lactams via the Castagnoli–Cushman reaction†
Anastasia
Lepikhina
,
Olga
Bakulina
,
Dmitry
Dar'in
and
Mikhail
Krasavin
*
Saint Petersburg State University, Saint Petersburg, 199034 Russian Federation. E-mail: m.krasavin@spbu.ru; Fax: +7 812 428 6939; Tel: +7 931 3617872
First published on 31st August 2016
Abstract
The first solvent-free protocol for the atom-economical Castagnoli–Cushman reaction is reported. It substantially broadens the reaction scope with respect to achievable substitution patterns around the privileged lactam cores compared to the traditional reaction format employing aromatic hydrocarbon solvents. The convenient product isolation protocol involves the use of aqueous base and acid solutions.
The formal cycloaddition reaction of imines (generated in situ or in a separate chemical step) with dicarboxylic acid anhydrides containing α-protons, known as the Castagnoli–Cushman reaction (CCR),1 provides a remarkably facile and, often, diastereoselective2 access to polysubstituted γ- and δ-lactams.3 The latter are exceptionally useful, privileged4 motifs in drug design considering the plethora of pharmacological effects reported for small molecules containing these heterocyclic cores.5 In addition, the possibility of independently varying the reaction components offered by this reaction (and multicomponent reactions in general), allows pre-meditating further reactions within the product of the multicomponent step and thus quickly building up skeletal complexity.6 One of the prominent features of this formally three-component process is its outstanding atom-economy:7 indeed, all atoms originating from the aldehyde, primary amine and cyclic anhydride components8 become incorporated into the product structure, except for the condensation water formed during the formation of the Schiff base (Scheme 1).
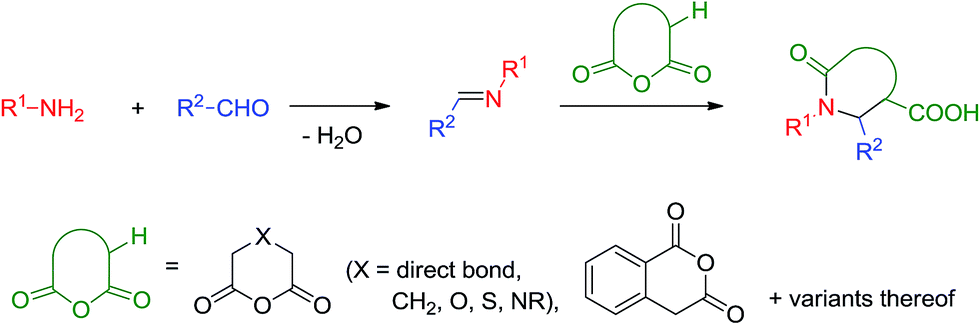 | ||
| Scheme 1 The Castagnoli–Cushman reaction. | ||
Considering that the crucial philosophical criterion of green chemistry, atom economy, is clearly fulfilled in the CCR, it is surprising that this important reaction has not been considered a tool for sustainable synthetic chemistry.9 A possible reason for that could be that the CCR is most often conducted in highly boiling aromatic hydrocarbon solvents (such as toluene or xylenes), while in many cases, chromatographic isolation of the reaction products is required.2 This leads to a significant production of waste and certainly limits the environmental friendliness of the process. This consideration prompted us to investigate the opportunity to develop a new format for the CCR which would align it with the spirit of green chemistry.10
Another prominent multicomponent process, the Ugi reaction of an amine, a carbonyl compound (the two forming a Schiff base), a carboxylic acid and an isocyanide (leading to a diamide adduct in a simplest case11) is not only feasible but proceeds with higher rates and produces cleaner product mixtures when conducted in water.12 This was a significant finding not only from the green chemistry13 but also from the general chemical reactivity perspective. Indeed, the Ugi reaction involves two hydrolytically prone components – the Schiff base (which can be hydrolyzed back to the primary amine and the carbonyl components) and the isocyanide (which can be hydrated to form the respective formamide, especially in presence of a carboxylic acid13) and yet the rates of potential side-reactions with water turn out to be lower than the principal multicomponent reaction course. Initially, we became inspired by this example and attempted conducting the CCR in aqueous medium. However, it quickly became apparent that the hydrolysis of the cyclic anhydride component in this case proceeded faster than the desired formal cycloaddition and the target lactam product did not form. At the same time, we had accidentally noted (in preliminary experiments that led to successful employment of thiodiglycolic anhydride in the CCR, X = S in Scheme 1(ref. 8)) that some reaction visibly occurred on neat mixing an imine and an anhydride components. This gave us an idea of conducting the CCR in the absence of any solvent which would not only eliminate the aromatic hydrocarbon solvent waste but also reduce the reaction volume and hence lower the energy requirements to achieve elevated reaction temperatures. In this communication, we report on a successful realization of this strategy using a set of N-arylidene anilines in combination with succinic and glutaric anhydrides (which are traditional inputs for the CCR1) as well as substituted versions of the latter.
The choice of the N-arylidene anilines as substrates (which were prepared in a separate step to improve product yield) was dictated primarily by the scarcity of the respective CCR products in the literature. Indeed, after the pioneering publication of Castagnoli,1a there has only been a handful of examples of 1,5-diaryl γ-lactams obtainable by the CCR of succinic anhydride15 (in some cases, this chemotype was accessed using alternative strategies16). Similarly, 1,6-diaryl δ-lactams amenable by the CCR are featured in only a few literature reports.17 Consequently, medicinal chemistry potential of these lactams remains to be unraveled beyond the disclosed analgesic,15a aromatase inhibitory15b,17d and urokinase receptor targeting anticancer15d activities. The reason for such compounds' being underrepresented in the literature could lie in the reluctance of the respective N-aryl imines to undergo the CCR. Indeed, according to the current mechanistic understanding,18 the CCR can proceed along two alternative mechanistic pathways (a: intermolecular N-acylation followed by intramolecular Mannich reaction or b: intermolecular Mannich reaction followed by intramolecular acylation) both of which include an acylation of the nitrogen atom in question (Scheme 2). Naturally, in aniline-derived imines, the nucleophilicity of that reactive center would be reduced and the reactivity of such substrates in the CCR would be retarded.
 | ||
| Scheme 2 Possible mechanistic pathways for the CCR (shown for succinic anhydride). | ||
In order to verify the viability of the solvent-free protocol and to determine the optimal temperature regimen for the CCR, neat equimolar mixtures of N-benzylidene aniline with succinic or glutaric anhydrides were heated at various temperatures ranging from 90 °C to 170 °C and the presence of the desired lactams as well as the yields were determined by the integration of the characteristic 1H NMR signals corresponding to C2–H of cis- and trans-isomers of the lactam products (the trans-isomer always being in majority17a) relative to n-tetradecane internal standard (see ESI†).
As can be seen from the results presented in Table 1, full conversions of the imine 1 (obtainable, in turn, by several green protocols from the respective aniline and aldehyde19) were achieved at temperatures above 130 °C and, to our delight, the signals corresponding to the desired product were present in the spectrum of the reaction mixture. The optimum product yield was observed at 150 °C and no significant improvement was evident at 170 °C. Therefore, the temperature of 150 °C was used uniformly throughout this study as shown in Scheme 3 (heating to 170 °C was used occasionally as noted below, in order to drive up the conversion and product yield, which were lower if the reaction was run at 150 °C).
|
|
||||||
|---|---|---|---|---|---|---|
| Temperature | n = 1 | n = 2 | ||||
| Conversion of the imine (%) | 1H NMR yield, % (trans + cis) | Conversion of the imine (%) | 1H NMR yield, % (trans + cis) | |||
| 90 °C | ∼50 | <20 | ∼40 | <20 | ||
| 110 °C | ∼60 | 22 | ∼50 | 24 | ||
| 130 °C | 100 | 36 | 98 | 42 | ||
| 150 °C | 100 | 48 | 100 | 65 | ||
| 170 °C | 100 | 50 | 100 | 66 | ||

 | ||
| Scheme 3 Solvent-free CCR of imines 1 with cyclic anhydrides 2–5 investigated in this work (the major, trans-isomer is shown for products 6–9). | ||
Reactions with succinic anhydride (2) all led to the formation of the desired products 6a–l (Table 2). After the heating of the neat mixture of the imine and succinic anhydride finished and the reaction was cooled down to ambient temperature, a glassy solid mixture was obtained. To that mixture, 10% aqueous KHCO3 (regarding the choice of the base – vide infra) was added and the solid mass gradually dissolved on overnight stirring, thus separating carboxylate-containing products from the insoluble by-products. The cloudy solution was filtered through a pad of Celite in order to further remove the latter and the filtrate was acidified causing the desired product of varying purity to precipitate. The isolated yield of 6a corresponded very well to the yield determined in the initial 1H NMR experiments (vide supra). Using this simple environmentally friendly isolation protocol, compounds 6a, 6b and 6d were obtained as analytically pure mixtures of trans- and cis-isomers with clear preference for the former (crystallization of 6d from aqueous ethanol furnished pure trans-isomer), which is in line with the results usually obtained in the aromatic hydrocarbon solvents.2 In the majority of cases, unfortunately, the product mixtures were contaminated with varying amounts of succinic monoanilide 10.
| Compound | Product 6 | Isolated yield (%) | Trans/cis ratio |
|---|---|---|---|
a The yield and trans![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cis ratio obtained after crystallization of the crude product from aqueous ethanol after the basic/acidic workup.
b Pure trans-isomer was obtained after additional crystallization from aqueous ethanol.
c The yield and trans:cis ratio obtained after crystallization of the crude product from aqueous ethanol without the basic/acidic workup.
d Reaction was run at 170 °C. :cis ratio obtained after crystallization of the crude product from aqueous ethanol after the basic/acidic workup.
b Pure trans-isomer was obtained after additional crystallization from aqueous ethanol.
c The yield and trans:cis ratio obtained after crystallization of the crude product from aqueous ethanol without the basic/acidic workup.
d Reaction was run at 170 °C.
|
|||
| 6a |

|
50 | 7:1 |
| 6b |

|
50 | 5:1 |
| 6c |

|
40a | 7:1 |
| 6d |

|
35 | 7:1b |
| 6e |

|
35a | 9:1a |
| 6f |

|
24a | 9:1a |
| 6g |

|
36a | 7:1a |
| 6h |

|
28a | 10:1a |
| 50c | 12:1c |
||
| 6i |

|
37a | 10:1a |
| 54c | 11:1c |
||
| 6j d |

|
37a | 10:1a |
| 6k |

|
37a | 10:1a |
| 6l |

|
35a | 6:1a |
| 63c | 7:1c |
||
The formation of 10 can be rationalized as follows. According to the current mechanistic understanding, acylation of the imine intermediate leads to zwitter-ionic species 11 that can productively cyclize to furnish the target compound 6. Alternatively, the iminium moiety can undergo an intramolecular trapping with carboxylate anion to give rise to cyclic aminal 12. The latter can, in principle, degrade back to 11 or it can eliminate aldehyde 14 and form succinimide 13 which can be hydrolyzed, on basic workup, to the contaminant 10 (Scheme 4). Some observations speak for the correctness of such mechanistic interpretation. Firstly, the use of more basic aqueous K2CO3 solution for product isolation led to much higher content of 10 in the isolated product 6. Secondly, 1H NMR analysis of the crude product prior to treatment with the aqueous base solution clearly displays characteristic signals corresponding to the aldehyde proton (δ ∼ 10.0 ppm) and the succinimide bis-methylene bridge (δ ∼ 2.9 ppm).
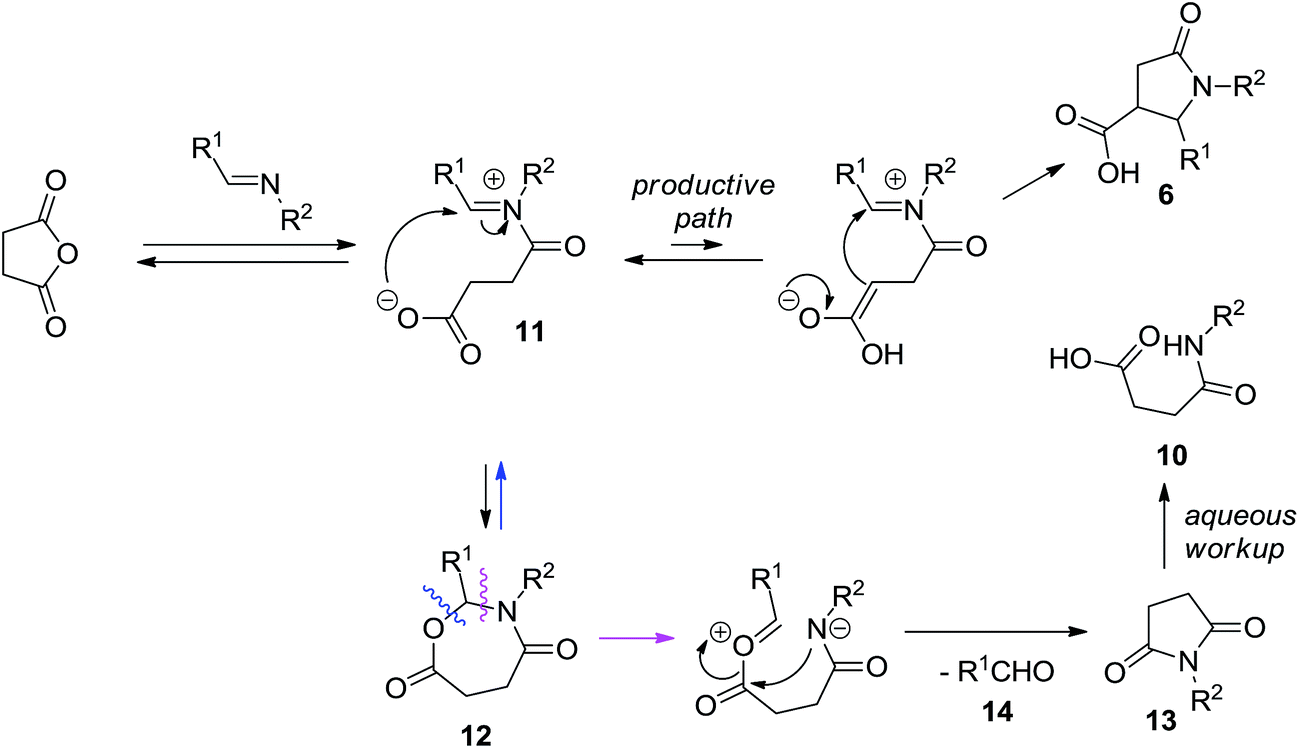 | ||
| Scheme 4 Possible competing reaction pathways rationalizing the formation of the by-products observed after the basic reaction workup. | ||
In all cases where the material precipitating on acidification of the basic solution of 6 was contaminated with 10, additional crystallization from aqueous ethanol furnished pure products 6 in modest yields and pronounced trans-selectivity. In some cases (6h, 6i, 6l), basic/acidic workup step could be omitted and crystallization from aqueous ethanol gave analytically pure products with significant improvement of the yield.
Employment of glutaric anhydride (3) as well as its 3,3-disubstituted variant (4–5) in the solvent-free CCR followed by the environmentally friendly basic/acidic aqueous isolation protocol described above, resulted in significantly improved product yields and purities while additional crystallization from aqueous ethanol was required in only one case, 7g (Table 3). Such a satisfactory result was likely due to lower likelihood of the reaction proceeding along the non-productive path as the formation of an 8-membered analog of postulated cyclic adduct 12 would be much less favored (however, contamination with glutaric monoanilide akin to 10 was still quite noticeable for 7g which mandated additional crystallization). Reactions involving 4 and 5 required higher temperature (170 °C) for better yields. This was likely due to much more sterically demanding situation around the α-position in these anhydrides, which is involved in the cyclization, rather than to possible Thorpe–Ingold effect.20 To the best of our knowledge, compounds 8a–d and 9a–d are the first examples of this type of highly sterically congested Castagnoli–Cushman-type δ-lactams.
| Compound | Product 7–9 | Isolated yield (%) | Trans/cis ratio |
|---|---|---|---|
|
a Pure trans-isomer was obtained after additional crystallization from aqueous ethanol.
b The yield and trans:cis ratio obtained after crystallization of the crude product from aqueous ethanol after the basic/acidic workup.
c Reaction was run at 170 °C.
d The yield and trans:cis ratio obtained after crystallization of the crude product from aqueous ethanol without the basic/acidic workup.
|
|||
| 7a |

|
67 | 6:1a |
| 7b |

|
65 | 7:1a |
| 7c |

|
72 | 8:1 |
| 7d |
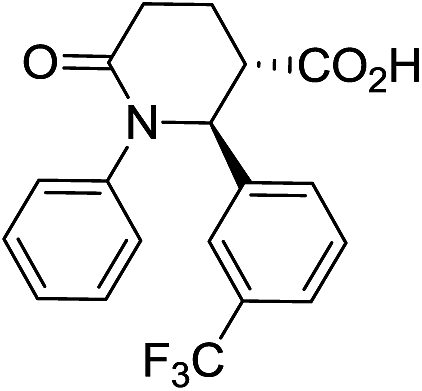
|
66 | 6:1 |
| 7e |
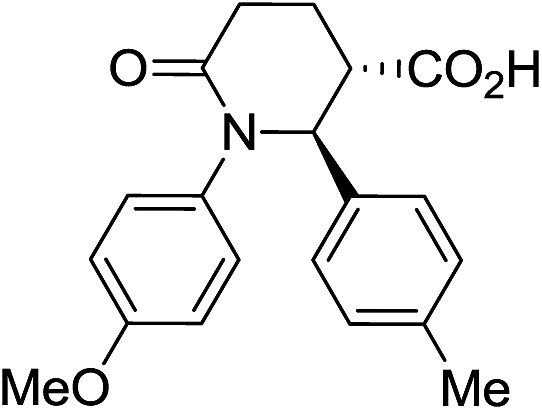
|
70 | 6.5:1 |
| 7f |
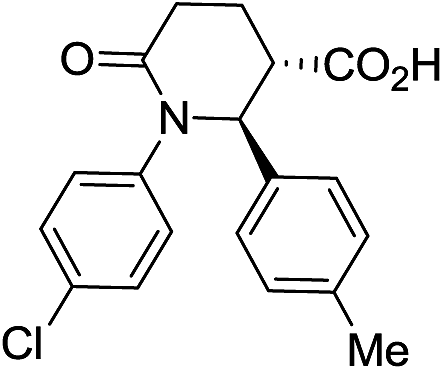
|
64 | 7:1 |
| 7g |

|
51b | 8:1a,b |
| 7h c |

|
74 | 6:1a |
| 7i |
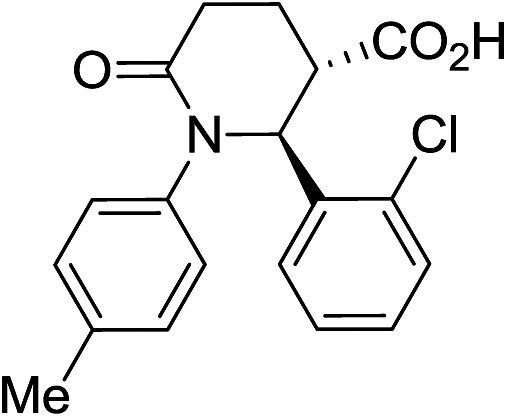
|
80 | 7:1a |
| 7j |

|
64 | 4:1 |
| 7k |

|
72 | 6:1 |
| 7l |
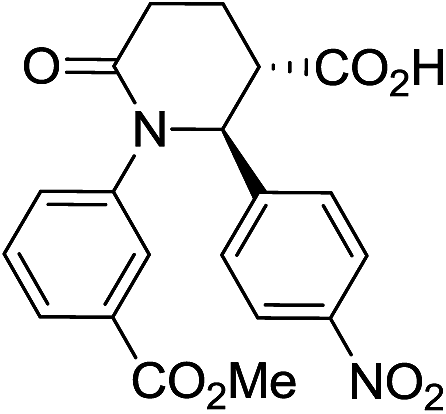
|
56 | 6:1a |
| 7m |

|
59d | Only trans |
| 7n |

|
89 | 5:1 |
| 7o |
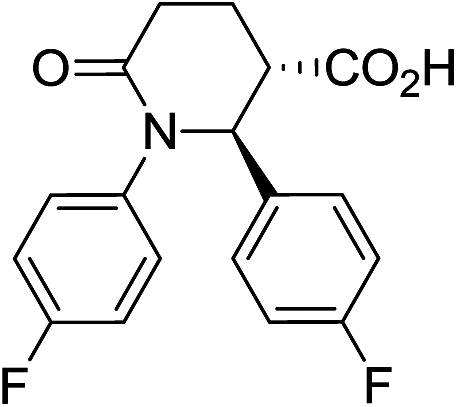
|
73 | 6:1 |
| 8a c |
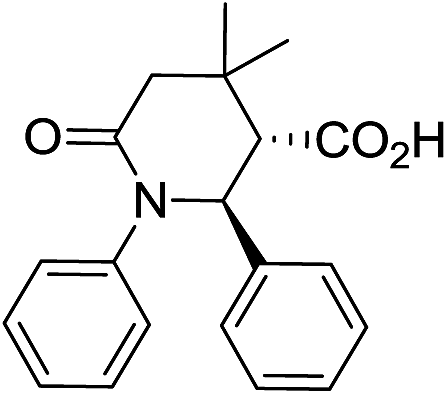
|
69 | 4:1a |
| 8b c |
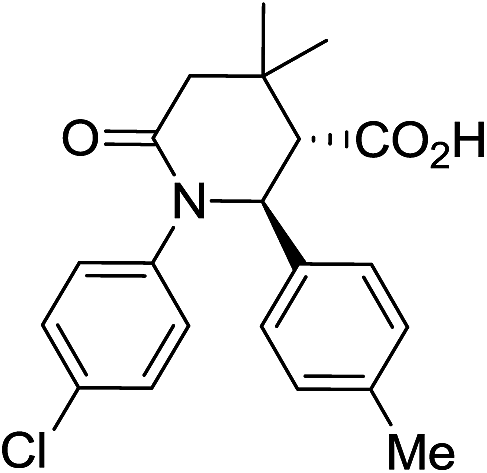
|
36 | 5:1 |
| 8c c |
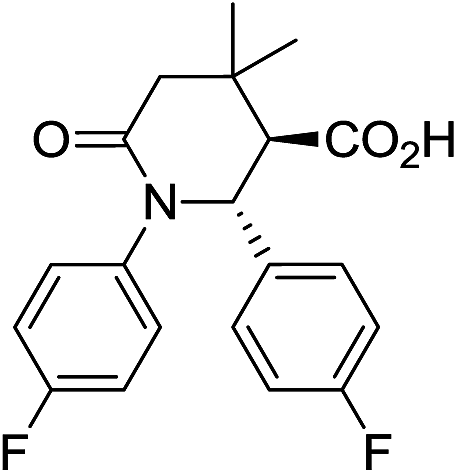
|
58 | 4:1 |
| 8d c |

|
50 | 7:1a |
| 9a c |
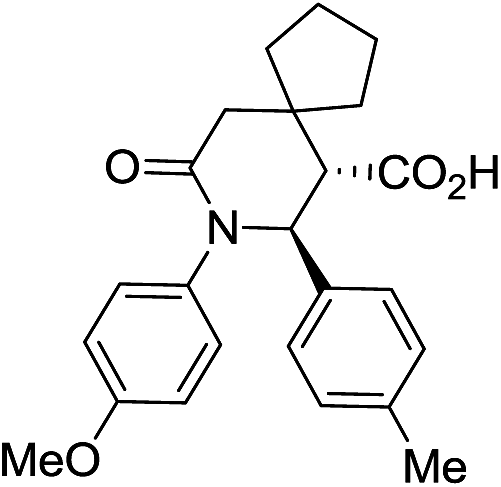
|
63 | 4:1a |
| 9b c |
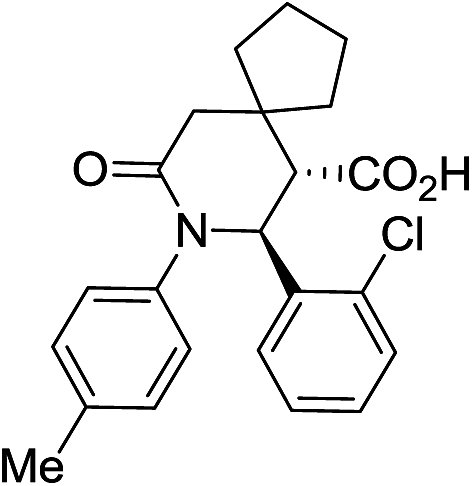
|
60 | 3:1 |
| 9c c |

|
30 | 2:1 |
| 9d c |

|
82 | 5:1 |
Notably, reactions summarized in Tables 2 and 3 appear to have a rather broad scope with respect to substituents in the aromatic groups originating from either the aldehyde or the aniline. In fact, the scarce examples of similarly diaryl-substituted CCR-derived lactams reported in the literature14–16 only include electron-neutral and electron-rich aromatic groups. In this study, similar reaction yields were obtained for diverse substitution patterns, including electron-withdrawing substituents in either portion of the imine partner (or even both portions as in 7l). We attempted to reproduce most striking examples of this sort (7d, 7h and 7l) in refluxing toluene and observed no formation of the respective products on prolonged (up to 72 h) reaction times. Therefore, the green, solvent-free protocol reported herein also enables the CCR reactions which are reluctant to proceed in solution.
In summary, we have reported the first solvent-free protocol for the synthesis of privileged γ- and δ-lactams using the Castagnoli–Cushman reaction which is not only environmentally friendly but also significantly broadens the scope of this important atom-economical reaction with respect to the accessible substitution patterns.
Acknowledgements
We gratefully acknowledge support from the Russian Scientific Fund (Project Grant 14-50-00069). NMR studies, X-ray analysis and Mass Spectrometry studies were performed at the Research Center for Magnetic Resonance, the Research Center for X-ray Diffraction Studies and the Center for Chemical Analysis and Materials Research of Saint-Petersburg State University.Notes and references
- (a) N. Castagnoli, J. Org. Chem., 1969, 34, 3187 CrossRef CAS PubMed; (b) M. Cushman and N. Castagnoli, J. Org. Chem., 1973, 38, 440 CrossRef CAS PubMed.
- M. González-López and J. T. Shaw, Chem. Rev., 2009, 109, 164 CrossRef PubMed.
- Recent reports on γ- and δ-lactam synthesis: (a) L. Deng, T. Xu, H. Li and G. Dong, J. Am. Chem. Soc., 2016, 138, 369–374 CrossRef CAS PubMed; (b) K. Kim and S. H. Hong, J. Org. Chem., 2015, 80, 4152–4156 CrossRef CAS PubMed; (c) V. Pharikronburee, T. Punirun, D. Soorukram, C. Kuhakarn, P. Tuchinda, V. Reutrakul and M. Pohmakotr, Org. Biomol. Chem., 2013, 11, 2022–2033 RSC.
- M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347 CrossRef CAS PubMed.
- K. S. Martin, M. J. DiMaso, J. C. Fettinger and J. T. Shaw, ACS Comb. Sci., 2013, 15, 356–362 CrossRef CAS PubMed.
- P. Sarnpitak and M. Krasavin, Tetrahedron Lett., 2014, 55, 2299 CrossRef CAS.
- B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 CrossRef CAS.
- D. Dar'in, O. Bakulina, M. Chizhova and M. Krasavin, Org. Lett., 2015, 17, 3930 CrossRef PubMed.
- R. C. Cioc, E. Ruijter and R. V. A. Orru, Green Chem., 2014, 16, 2958 RSC.
- Y. Gu, Green Chem., 2012, 14, 2091 RSC.
- A. Dömling and I. Ugi, Angew. Chem., Int. Ed. Engl., 2000, 39, 3168 CrossRef.
- M. C. Pirrung and K. Das Sarma, J. Am. Chem. Soc., 2004, 126, 444 CrossRef CAS PubMed.
- I. Kanizsai, S. Gyonfalvi, Z. Szakonyi, R. Illanpää and F. Fülöp, Green Chem., 2007, 9, 357 RSC.
- E. Ohta, M. M. Uy, S. Ohta, M. Yanai, T. Hirata and S. Ikegami, Biosci., Biotechnol., Biochem., 2008, 72, 1764 CrossRef CAS PubMed.
- (a) B. V. Shetty, A. McFadden and P. Hofer, US Pat., 4,476,311 1984Chem. Abstr., 1985, 102, 78717; (b) M. Tabcheh, M. Baroudi, F. Elomar, A. Elzant, M. Elkhatib and V. Rolland, Asian J. Chem., 2006, 18, 1771 CAS; (c) J. Wei and J. T. Shaw, Org. Lett., 2007, 9, 4077 CrossRef CAS PubMed; (d) T. Mani, D. Liu, D. Zhou, L. Li, W. E. Knabe, F. Wang, K. Oh and S. O. Meroueh, ChemMedChem, 2013, 8, 1963 CrossRef CAS PubMed.
- (a) M. Pohmakotr, N. Yotapan, P. Tuchinda, C. Kuhakarn and V. Reutrakul, J. Org. Chem., 2007, 72, 5016 CrossRef CAS PubMed; (b) M. Pohmakotr, N. Yotapan, P. Tuchinda, C. Kuhakarn and V. Reutrakul, Tetrahedron, 2007, 63, 4328 CrossRef CAS; (c) Z. Li, Y. Feng, Z. Li and L. Jiang, Synlett, 2014, 25, 2899 CrossRef CAS.
- (a) D. Dar'in, O. Bakulina, S. Nikolskaya, I. Gluzdikov and M. Krasavin, RSC Adv., 2016, 6, 49411 RSC; (b) M. J. DiMaso, K. M. Snyder, F. De Souza Dernandes, O. Pattawong, D. Q. Tan, J. C. Fettinger, P. H.-Y. Cheong and J. T. Shaw, Chem.–Eur. J., 2016, 22, 4794 CrossRef CAS PubMed; (c) M. Baroudi, J. Robert and C. Luu-Duc, Heterocycl. Commun., 1996, 2, 255–260 CrossRef CAS; (d) M. Baroudi, J. Robert and C. Luu-Duc, J. Steroid Biochem. Mol. Biol., 1996, 57, 73 CrossRef CAS PubMed.
- M. Krasavin and D. Dar'in, Tetrahedron Lett., 2016, 57, 1635 CrossRef CAS.
- (a) J. S. Bennett, K. L. Charles, M. R. Miner, C. F. Heuberger, E. J. Spina, M. F. Bartels and T. Foreman, Green Chem., 2009, 11, 166 RSC; (b) V. K. Rao, S. S. Reddy, B. S. Krishna, K. R. M. Naidu, C. N. Raju and S. K. Ghosh, Green Chem. Lett. Rev., 2010, 3, 217 CrossRef CAS; (c) M. G. Dekamin, M. Azimoshan and L. Ramezani, Green Chem., 2013, 15, 811 RSC.
- S. M. Bachrach, J. Org. Chem., 2008, 73, 2466 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1470615, 1470616 and 1470618. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra19196g |
| This journal is © The Royal Society of Chemistry 2016 |