
Thermochemical conversion of CO2 into CH4 using oxygen deficient NiFe2O4−δ with unique selectivity†
Zhifu
Liu,  *a
*a
*a
Abstract
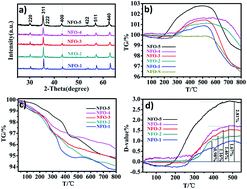
NiFe2O4 nanoparticles with a high concentration of oxygen vacancies were synthesized via a solvothermal route. The morphology and oxygen vacancies of the catalyst was changed by adjusting the ratio of ethylene glycol (EG) in the mixture solvent. Water was used as a hydrogen source, the thermal catalytic performance of NiFe2O4 was up to 357.6 μmol g−1 of CH4 evolution under low temperature conditions. These findings may further broaden the horizon for CO2 reduction using the oxygen nonstoichiometry strategy.
 Please wait while we load your content...
Please wait while we load your content...

