
Role of Fe in the oxidation of methanol electrocatalyzed by Ni based layered double hydroxides: X-ray spectroscopic and electrochemical studies

Abstract
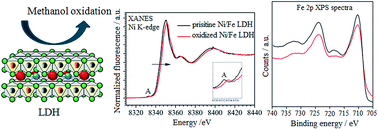
Ni/Al and Ni/Fe layered double hydroxides (LDHs) were electrosynthesized on Pt electrodes to be used as catalysts for the development of methanol fuel cells. The electrochemical characterization and the electrocatalytic activity of the two LDHs towards methanol electro-oxidation in alkaline conditions were investigated. Furthermore, the role of Fe on the electronic and structural properties of the LDHs was investigated performing X-ray Absorption Spectroscopy (XAS) and X-ray photoelectron spectroscopy (XPS). By the means of these techniques the materials were studied just electrosynthesized and after potentiostatic oxidation. The percentage of Ni active sites in the Ni/Fe LDH was higher than that of Ni/Al LDH, leading to a more efficient catalytic effect towards methanol oxidation in terms of current recorded at any potential value. Furthermore, methanol oxidation occurred at a lower potential for the same current density in the case of Ni/Fe LDH. The electrocatalytic performance displayed by the Ni/Fe LDH suggested the occurrence of a synergic effect between Ni and Fe sites, even if Fe is not directly involved in the redox process, and this evidence was confirmed by XAS and XPS experiments.
 Please wait while we load your content...
Please wait while we load your content...

