
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper(I)-mediated synthesis of β-hydroxysulfones from styrenes and sulfonylhydrazides: an electrochemical mechanistic study†
Alexander O.
Terent'ev
 *ab,
Olga M.
Mulina
a,
Dmitry A.
Pirgach
ab,
Dmitry V.
Demchuk
a,
Mikhail A.
Syroeshkin
a and
Gennady I.
Nikishin
a
*ab,
Olga M.
Mulina
a,
Dmitry A.
Pirgach
ab,
Dmitry V.
Demchuk
a,
Mikhail A.
Syroeshkin
a and
Gennady I.
Nikishin
a
aN. D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky Prospect 47, Moscow, 119991, Russian Federation. E-mail: terentev@ioc.ac.ru; Fax: +7-499-1355328
bD. I. Mendeleev University of Chemical Technology of Russia, 9 Miusskaya square, Moscow, 125047, Russian Federation
First published on 14th September 2016
Abstract
Copper(I) halides were used as mediators in the synthesis of β-hydroxysulfones via the oxysulfonylation of styrenes using sulfonylhydrazides. The feature of the developed process lies in the combination of a copper(I) salt with oxygen—the stoichiometric oxidant. Copper(II) species are responsible for the oxidation of sulfonylhydrazides, they are generated in small amounts in the O2/Cu(I)/Cu(II) redox system, which is formed during the reaction. The combination of these three components enables one to obtain in the case of α-methylstyrenes only β-hydroxysulfones and in the case of α-unsubstituted styrenes, β-hydroxysulfones as the main products and β-ketosulfones as the by-products. With good yields β-hydroxysulfones were prepared by reduction of the reaction mixture containing both products β-hydroxysulfones and β-ketosulfones with NaBH4. An electrochemical study revealed that the Cu(I)/Cu(II) pair can serve as an effective mediator of β-hydroxysulfones formation via redox processes.
Introduction
β-Hydroxysulfones are of great interest as structural units of antifungal1 and antitumor2 compounds, they are known as intermediates in the synthesis of lactones3 and unsymmetrical alkenes.4 Traditionally β-hydroxysulfones are obtained through the nucleophilic addition of sulfinates to epoxides,5–8 reduction of β-ketosulfones9,10 and hydroxylation of α,β-unsaturated sulfones.11 Over the last few years, several oxidative strategies to prepare β-hydroxysulfones from olefins have been established. In these reports air and low thermally stable sulfinic acids12–14 in combination with O2 and PPh3 (Scheme 1, eqn (1))15 or sulfonylhydrazides with O2 and Fe(III) salts are required.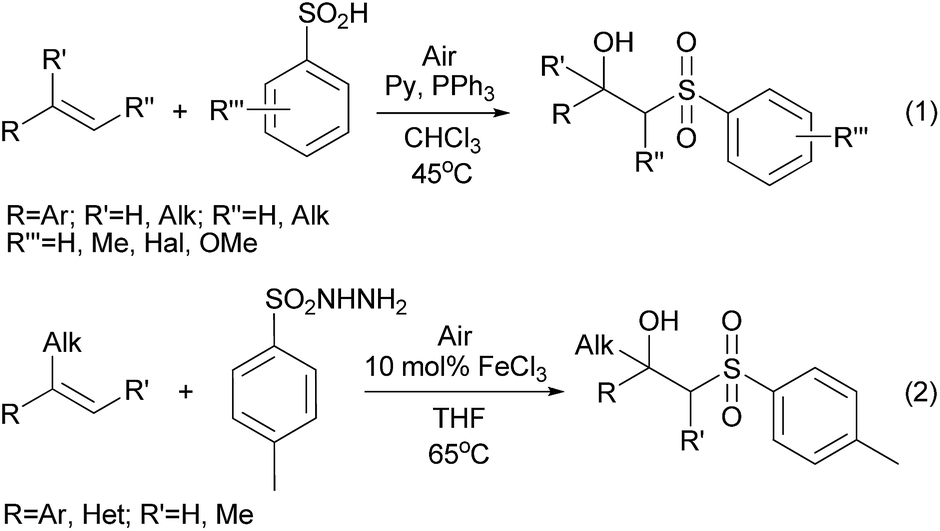 | ||
| Scheme 1 Recent works for the oxysulfonylation of styrenes. | ||
The latter method was applied for the synthesis of structures containing the β-hydroxysulfone moiety predominantly at a tertiary carbon atom, which cannot be further oxidized (Scheme 1, eqn (2)).16
For the sulfonylation of unsaturated compounds without additional insertion of oxygen into the molecule, a number of oxidants have been exploited: Cu(OAc)2,17,18 CAN,19 NBS,20 K2S2O8,21 peroxides,22 I2/TBHP23 and TBAI/TBHP24,25 systems. It is well-known that oxygen is an ideal environmentally friendly oxidant, which offers fascinating industrial and academic prospects. In oxidative transformations, in most cases, it is used in combination with transition metals salts and complexes.26–29
In this context, we have disclosed a process for the oxysulfonylation of styrenes utilizing sulfonylhydrazides in the presence of a O2/Cu(I) system, leading to β-hydroxysulfones. During the reaction the O2/Cu(I)/Cu(II) system is formed with a small amount of Cu(II) as confirmed by the near absence of the specific colour of Cu(II) species. As a result, β-hydroxysulfones 3 as main products and β-ketosulfones 4 as by-products are formed (Scheme 2).
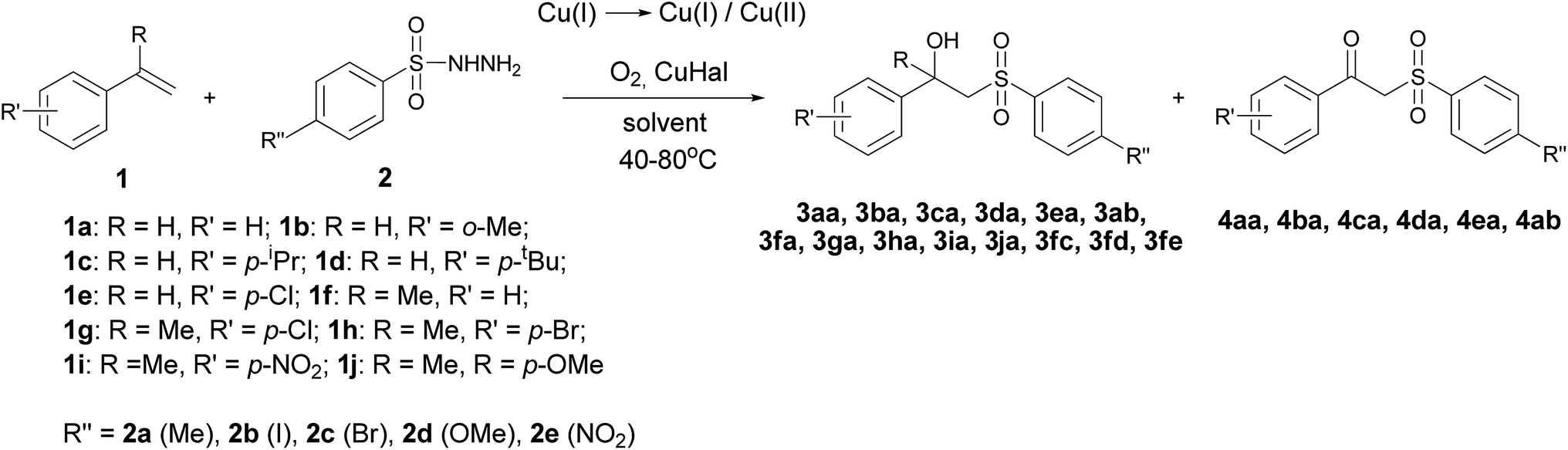 | ||
| Scheme 2 The oxysulfonylation of styrenes 1a–1j using sulfonylhydrazides 2a–2e (in the codification of 3 and 4 the first letter index refers to the styrene 1 moiety, the second letter index to the hydrazide 2 moiety). | ||
Results and discussion
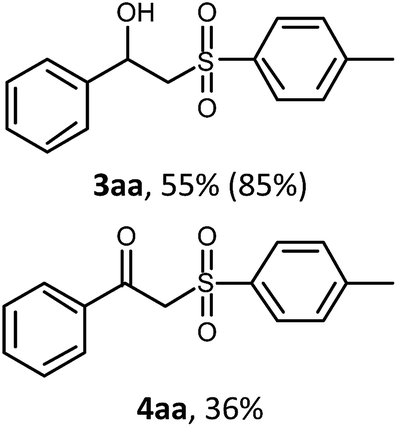
The synthesis of β-hydroxysulfones 3aa–3fe and β-ketosulfones 4aa–4ab from styrenes 1a–1j with the use of sulfonylhydrazides 2a–2e was conducted in CH3CN, CH3CN–H2O, THF and THF–H2O using a O2/Cu(I)/Cu(II) redox system. This system was a result of the transformation of CuCl, CuBr and CuI under aerobic conditions (Scheme 2).Our preliminary studies were focused on the reaction of styrene 1a with sulfonylhydrazide 2a, leading to the formation of 1-phenyl-2-tosylethanol 3aa and 1-phenyl-2-tosylethanone 4aa. We examined the influence of the Cu(I) salt counter-ion, oxygen source (air oxygen or 98% oxygen) and solvent type (Table 1) on the yield of 3aa.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Time (h) | Ratio (mole Cu(I)/mole 1a) | Oxygen source | Solvent | Yield 3aab (%) | Yield 4aab (%) | Total yield 3aa and 4aab (%) |
a General procedure: to a solution of styrene 1a (300 mg, 2.88 mmol) in 25 mL of (CH3CN–H2O (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), CH3CN, THF, THF–H2O (5:1)), the Cu(I) salt (0.58–14.4 mmol, molar ratio 0.2–5 mol of salt/mol 1a) and sulfonylhydrazide 2a (537 mg, 2.88 mmol, molar ratio 1 mol 2a/mol 1a) were added. The mixture was stirred for 7 h at 40 °C.
b The yield was determined by 1H NMR using 1,4-dinitrobenzene as an internal standard, the isolated yield after reduction with NaBH4 is reported in the parentheses.
c 7 h at 40 °C, then 12 h at 20–25 °C.
d 7 h at 80 °C, then 12 h at 20–25 °C. :1), CH3CN, THF, THF–H2O (5:1)), the Cu(I) salt (0.58–14.4 mmol, molar ratio 0.2–5 mol of salt/mol 1a) and sulfonylhydrazide 2a (537 mg, 2.88 mmol, molar ratio 1 mol 2a/mol 1a) were added. The mixture was stirred for 7 h at 40 °C.
b The yield was determined by 1H NMR using 1,4-dinitrobenzene as an internal standard, the isolated yield after reduction with NaBH4 is reported in the parentheses.
c 7 h at 40 °C, then 12 h at 20–25 °C.
d 7 h at 80 °C, then 12 h at 20–25 °C.
|
|||||||
| 1 | 7 | CuBr (2) | Air | CH3CN–H2O | 28 | 14 | 42 |
| 2 | 7 | CuCl (2) | Air | CH3CN–H2O | 25 | 15 | 40 |
| 3 | 7 | CuI (2) | Air | CH3CN–H2O | 17 | 10 | 27 |
| 4c | 7 + 12 | CuBr (2) | Air | CH3CN–H2O | 38 (61) | 27 | 65 |
| 5d | 7 + 12 | CuBr (2) | Air | CH3CN–H2O | 30 | 16 | 46 |
| 6 | 7 + 12 | CuBr (0.2) | Air | CH3CN–H2O | 10 | 5 | 15 |
| 7 | 7 + 12 | CuBr (5) | Air | CH3CN–H2O | 36 | 17 | 53 |
| 8 | 7 + 12 | CuBr (2) | Air | CH3CN | 20 | 12 | 32 |
| 9 | 7 + 12 | CuBr (2) | Air | THF | 28 | 13 | 41 |
| 10 | 7 + 12 | CuBr (2) | Air | THF–H2O | 18 | 10 | 28 |
| 11 | 7 + 12 | CuBr (2) | O2 | CH3CN–H2O | 55 (85) | 36 | 91 |
| 12 | 7 + 12 | CuBr (0.2) | O2 | CH3CN–H2O | 15 | 8 | 23 |
| 13 | 7 | CuBr (2) | O2 | CH3CN–H2O | 43 (71) | 35 | 78 |
| 14d | 7 + 12 | CuBr (2) | O2 | CH3CN–H2O | 50 (77) | 30 | 80 |

Entries 1–3 indicated that among the copper(I) halides (CuBr, CuCl and CuI), the use of CuBr afforded the highest total yield of oxysulfonylation products and the yield of the desired product 3aa after 7 h. When the reaction was performed for a more prolonged time (entry 4) the yield of 3aa reached 38%. Heating the reaction mixture for the first 7 h to 80 °C (entry 5) didn't increase the yield of 3aa. Decreasing (entry 6) or increasing (entry 7) the molar ratio of CuBr per mol of 1a in comparison with the previous entries resulted in a reduced yield of the desired product. Employing CH3CN, THF or THF–H2O (5:1) in place of CH3CN–H2O (5:1) negatively influenced the reaction efficiency (entries 8–10). In entry 11, air oxygen was replaced with 98% oxygen and as a result the yield of 3aa was improved to 55%, the total yield of oxysulfonylation products in this case reached 91%. Attempts failed (entries 12–14) to increase the yield of desired product through modification of the molar ratio of CuBr per mol of 1a, temperature and reaction time when compared to the conditions of entry 11, in which the best result was obtained; the total yield of 3aa and 4aa in these experiments didn't exceed 80%.
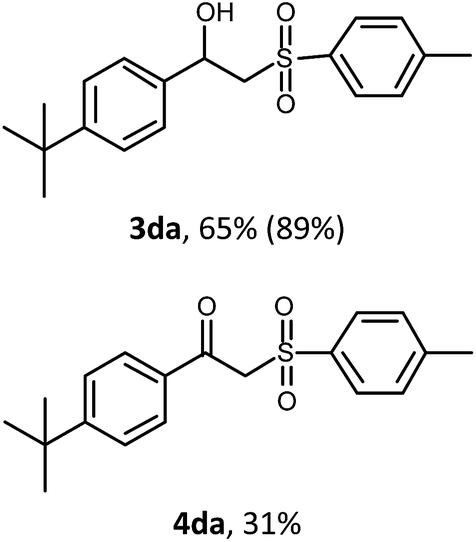
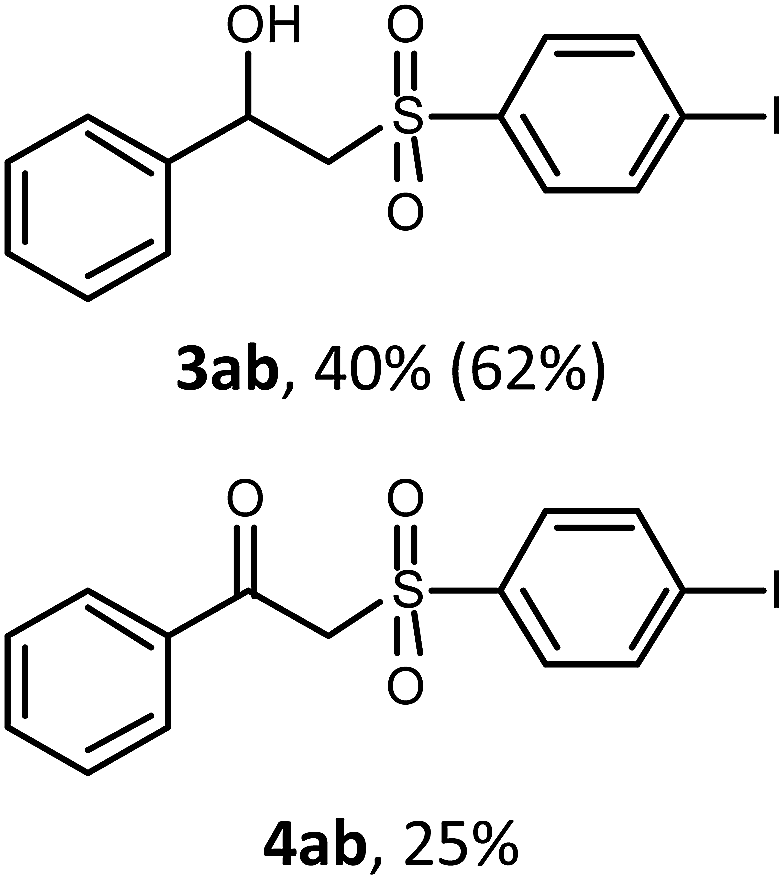
With the optimized reaction conditions in hand (entry 11, Table 1), the scope of the copper-mediated oxysulfonylation reaction was investigated. A number of β-hydroxysulfones 3aa–3ab were formed in 32–65% yield with β-ketosulfones 4aa–4ab observed as the by-products of the reaction in 18–33% yield (Table 2).
| a The yield was determined by 1H NMR using 1,4-dinitrobenzene as an internal standard; the isolated yield after reduction with NaBH4 is reported in the parentheses. | |
|---|---|
|

|

|
|

|
|
In all the examples, hydroxysulfone was predominantly formed independently of the properties of the substituents on the benzene ring. In most cases, the molar ratio of β-hydroxysulfone 3/ketosulfone 4 was 2:1. It is well-known that ketones can be easily reduced into their corresponding alcohols.30–32 That's why in order to transform the β-ketosulfones 4 by-products into the desired β-hydroxysulfones 3 we filtered the reaction mixture from the CuBr after the reaction was complete and then carried out the reduction of the ketosulfones using NaBH4 (Scheme 3). As a result, the desired β-hydroxysulfones 3 were obtained in a 52–89% overall yield over two steps (Table 2 and entries 4, 11, 13, 14 in Table 1).
 | ||
| Scheme 3 The reduction of β-ketosulfones using NaBH4. | ||
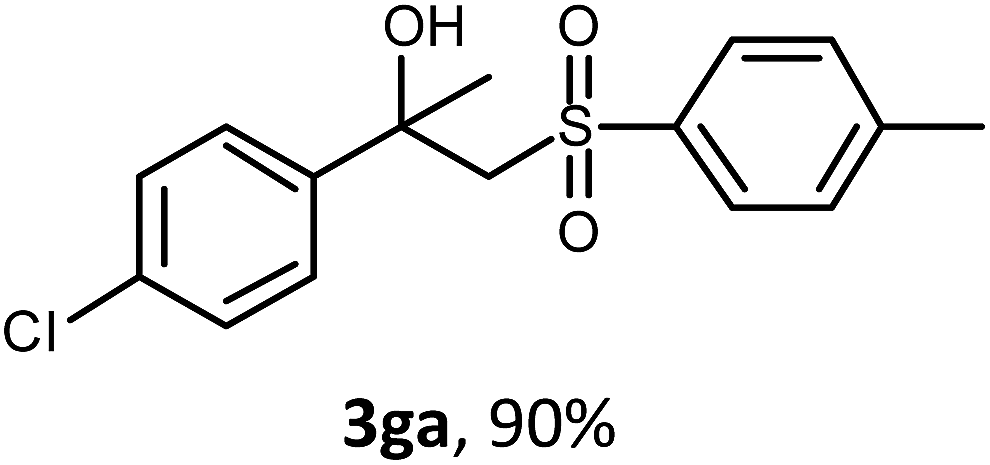
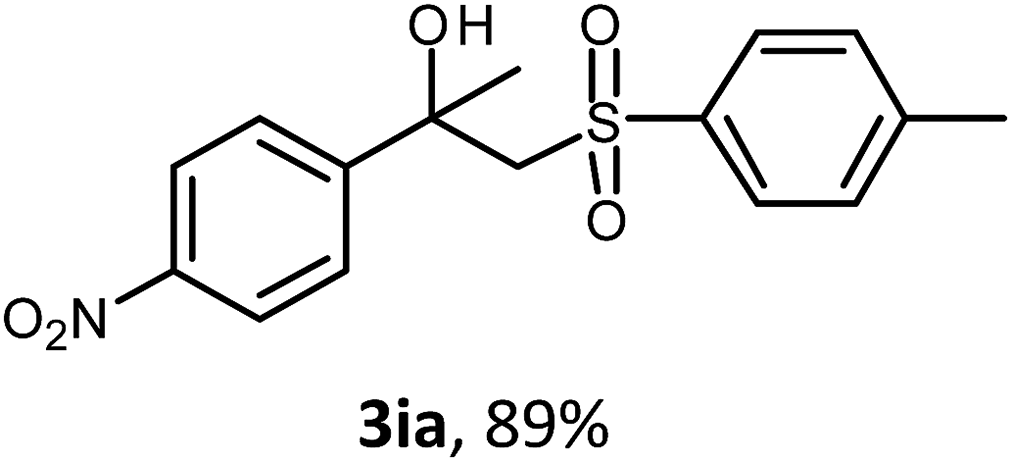
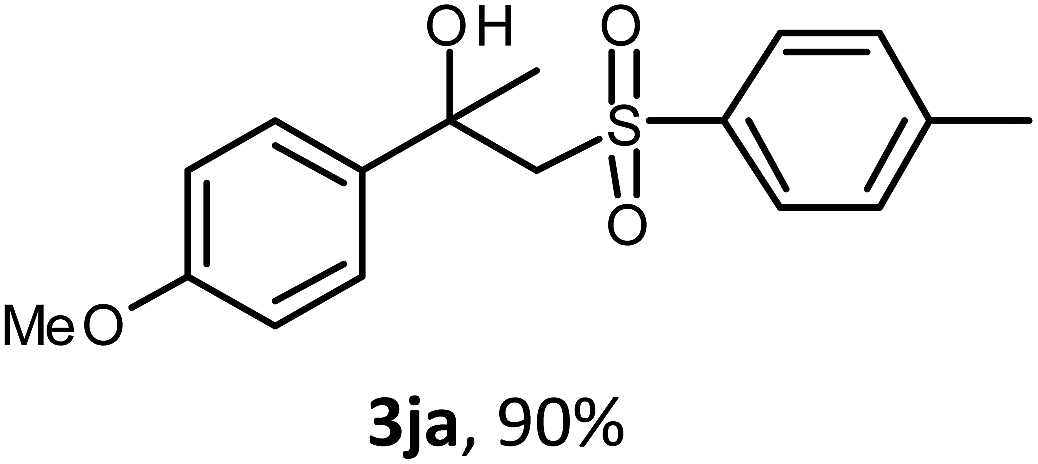
The oxysulfonylation reaction was also examined by applying of α-methylstyrenes (Table 3). A variety of α-methylstyrenes bearing either electron withdrawing or electron donating substituents on the aryl ring worked well under the conditions of entry 11 (Table 1) and the target β-hydroxysulfones were formed in most cases in good yield. It is important to note that in the reactions of methylsulfonylhydrazide with styrene 1a and octene-1 or cyclohexene with sulfonylhydrazide 2a the oxysulfonylation products were not observed in measurable yield.
| a Isolated yield. | |
|---|---|

|
|

|
|
|

|
|

|
The structures of all the synthesized hydroxysulfones 3 and ketosulfones 4 were confirmed by 1H and 13C NMR spectroscopy, elemental analysis, HRMS and IR spectroscopy.
Proposed reaction mechanism
To study the plausible reaction mechanism, an investigation of the redox properties of oxygen, styrene 1a, p-toluenesulfonylhydrazide 2a and CuBr in CH3CN–H2O (5:1) with the use of cyclic voltammetry (CV) was carried out. Tetrabutylammonium perchlorate was chosen as a supporting electrolyte. The obtained CV curves are shown in Fig. 1.
 | ||
| Fig. 1 The CV curves obtained for 2.0 mmol L−1 solutions of CuBr (red), p-toluenesulfonylhydrazide 2a (blue), styrene 1a (black) and aerated supporting solution (grey) in 0.1 M Bu4NClO4 in CH3CN–H2O (5:1) on a working glassy-carbon electrode (d = 1.7 mm) at a scan rate of 100 mV s−1. | ||
The oxidizing properties of oxygen are evidenced in reducing at the relatively early potentials, the peak on the CV curve with potential −1.10 V was responsible for its reduction. The chemically irreversible oxidation of styrene 1a takes place in the far region with a maximum at 1.90 V and runs into the discharge of the background. A chemically irreversible peak at 1.35 V was responsible for the oxidation of p-toluenesulfonylhydrazide 2a. Therefore, we can conclude that its oxidation goes rather more easily than the oxidation of styrene.
A chemically and electrochemically reversible peak at E1/2 = 0.55 V corresponds to the oxidation of Cu(I) into Cu(II), which takes place in the potential range between oxygen reduction and p-toluenesulfonylhydrazide 2a oxidation. This means that under experimental conditions the Cu(I)/Cu(II) couple can serve as an effective mediator of p-toluenesulfonylhydrazide 2a oxidation using oxygen.
On the basis of the obtained experimental data and previous studies of reactions proceeding through the generation of S-centered radicals from sulfonylhydrazides,33–35 we proposed the pathway of the oxysulfonylation process (Scheme 4). Cu(I) ions are oxidized to Cu(II)36–40 in the presence of oxygen, as confirmed by numerous kinetic studies of this process.41–44 An almost colourless solution during the reaction is evidence for a small amount of Cu(II). Afterwards, as a result of the successive oxidation of hydrazide 2a under the action of Cu(II),16,33,34 oxygen or peroxyradical C, an S-centered tosyl radical A (Ts) is generated, which reacts with styrene 1a to form a C-centered benzyl radical B.16 In the next step, radical B is trapped with oxygen forming peroxyradical C. Then, after abstraction of an hydrogen atom from a hydrogen donor X–H (NH and CH) the peroxyradical C transforms into hydroperoxide D,45 which gives the main product 3aa after reduction.46–51 Alcohol 3aa can also be formed due to C-centered benzyl radical B oxidation by Cu(II) species to the intermediate cation E followed by its hydroxylation.52–54 Ketone 4aa is formed after the fragmentation of the species generated from the reaction of peroxyradical C with Cu(I) ions55 or oxygen.56–58
 | ||
| Scheme 4 A plausible oxysulfonylation mechanism using the example of the reaction of styrene 1a and p-toluenesulfonylhydrazide 2a. | ||
The fact that using of octene-1 and cyclohexene as starting reagents didn't lead to the formation of oxysulfonylation products can be explained by the low stability of the C-centered alkyl radical generated after addition of tosyl radical A to their double bonds.
Conclusions
In summary, we have demonstrated a novel copper-mediated oxysulfonylation of styrenes using sulfonylhydrazides for the synthesis of β-hydroxysulfones in 32–93% yield. In the case of α-unsubstituted styrenes, β-ketosulfones are formed as the by-products. Applying α-methylstyrenes in this methodology gives β-hydroxysulfones as single products in high yield. Moreover, using cyclic voltammetry, experimental data and previous reports, a plausible reaction pathway was proposed. Coupling of two starting reagents proceeds under the action of the O2/Cu(I)/Cu(II) redox system. The distinguishing feature of the work lies in the combination of oxygen and a copper(I) salt, which is oxidized to copper(II) on a limited scale, which makes it possible to obtain β-hydroxysulfones as the main products.Experimental
NMR spectra were recorded on a Bruker Avance II 300 MHz instrument. Chemical shifts are measured relative to the residual solvent peaks as an internal standard set to δ 7.25 and δ 77.0 (CDCl3), and δ 2.50 and δ 39.51 (DMSO-d6). IR spectra were recorded on a FT-IR spectrometer. High resolution mass spectra (HRMS) were measured using electrospray ionization (ESI).59 The measurements were carried out in positive ion mode (interface capillary voltage 4500 V); the mass ratio was from m/z 50 to 3000 Da; external/internal calibration was performed using an electrospray calibrant solution. Syringe injection was used for solutions in CH3CN (flow rate 3 µL min−1). Nitrogen was applied as a dry gas and the interface temperature was set at 180 °C. TLC analyses were carried out on standard silica-gel chromatography plates. Melting points were determined on a Kofler hot-stage apparatus. Chromatography was performed on silica gel (63–200 mesh).Vinylbenzene (1a), 1-methyl-2-vinylbenzene (1b), 1-tert-butyl-4-vinylbenzene (1d), 1-chloro-4-vinylbenzene (1e), isopropenyl-benzene (1f), p-toluenesulfonohydrazide (2a), 4-iso-propylbenz-aldehyde, 4-chloroacetophenone, 4-bromoacetophenone, 4-nitroacetophenone, 4-methoxyacetophenone, 4-iodobenzene-sulfonylchloride, 4-bromobenzenesulfonylchloride, 4-methoxy-benzenesulfonylchloride, 4-nitrobenzenesulfonylchloride, methyltri-phenylphosphonium bromide, t-BuOK, Na2SO4, tetra-butyl-ammonium perchlorate, CuBr, CH3CN, THF, CHCl3, CH2Cl2, MeOH, petroleum ether (PE, 40/70), ethyl acetate (EA) and hydrazine hydrate (64% w/w water solution of hydrazine) were purchased from commercial sources and were used as received. 1-Iso-propyl-4-vinylbenzene (1c), 1-chloro-4-isopropenylbenzene (1g), 1-bromo-4-isopropenylbenzene (1h), 1-nitro-4-isopropenylbenzene (1i) and 1-methoxy-4-isopropenylbenzene (1j) were synthesized via the Wittig reaction according to the literature.60 4-Iodobenzenesulfono-hydrazide (2b), 4-bromobenzenesulfonohydrazide (2c), 4-methoxy-benzenesulfonohydrazide (2d) and 4-nitrobenzenesulfonohydrazide (2e) were synthesized according to the literature.61
Cyclic voltammetry (CV) was implemented on an IPC-Pro computer-assisted potentiostat manufactured by Econix (scan rate error 1.0%; potential setting 0.25 mV). The experiments were performed in a 10 mL five-neck glass conic electrochemical cell with a water jacket for thermostatting. CV curves were recorded using a three-electrode scheme. The working electrode was a disc glassy-carbon electrode (d = 1.7 mm). A platinum wire served as an auxiliary electrode. A saturated calomel electrode was used as the reference electrode and was linked to the solution by a bridge with a porous ceramic diaphragm filled with background electrolyte. The tested solutions were thermostatted at 25 ± 0.5 °C. In a typical case, 5 mL of solution was utilized and the depolarizer concentration was 2 mmol L−1. The working electrode was polished before recording each CV curve.
Synthesis of styrenes 1c, 1g–1j
Following a literature procedure60 to a solution of methyltriphenylphosphonium bromide (14.3–19.1 g, 39.9–53.4 mmol) in THF (50 mL), t-BuOK (4.8–6.5 g, 43.1–57.7 mmol) was added with vigorous stirring under an Ar atmosphere for 10 min. The mixture was stirred for 1 h at room temperature. Then, the corresponding carbonyl compound (4-iso-propylbenzaldehyde, 4-chloroacetophenone, 4-bromoacetophenone, 4-nitroaceto-phenone or 4-methoxyacetophenone, 3.0 g, 15.1–20.2 mmol) was added. The mixture was stirred for 24 h at room temperature. Then, the mixture was diluted with CH2Cl2 (180 mL), washed with water (3 × 15 mL) and dried over Na2SO4. The solvent was removed under reduced pressure (10–20 Torr). Styrenes 1c, 1g–1j were isolated by chromatography on SiO2 with elution using PE–EA with a linear gradient of the latter from 0 to 10 vol%.Synthesis of sulfonylhydrazides 2b–2e
Following a literature procedure61 a THF (20 mL) solution containing the corresponding arylsulfonyl chloride (4-iodobenzene-sulfonylchloride, 4-bromobenzenesulfonylchloride, 4-methoxy-benzenesulfonylchloride, 4-nitrobenzenesulfonylchloride, 5.0 g, 16.5–24.2 mmol) was cooled using an ice-water bath to 5 °C. Hydrazine hydrate (64% w/w water solution of hydrazine, 2.1–3.0 g; 41.2–60.5 mmol) was slowly added with stirring. Then, the reaction mixture was stirred at 5 °C for 30 min, diluted with THF (20 mL), washed with water (3 × 5 mL) and dried over Na2SO4. The solvent was removed under reduced pressure (10–20 Torr) to give the pure products 2b–2e.General procedure 1. Preparation of 3aa and 4aa. Optimization of the reaction conditions for oxysulfonylation of styrene 1a with sulfonylhydrazide 2a (Table 1)
To a solution of styrene 1a (300 mg, 2.88 mmol) in 25 mL of (CH3CN–H2O (5:1), CH3CN, THF, THF–H2O (5:1)), Cu(I) salt (0.58–14.4 mmol, molar ratio 0.2–5 mol of salt/mol 1a) and p-toluenesulfonylhydrazide 2a (537 mg, 2.88 mmol, molar ratio 1 mol 2a/mol 1a) were added. The mixture was stirred in the air or an oxygen atmosphere for 7 h at 40 °C, for 7 h at 80 °C, then for 12 h at room temperature.
Treatment of the reaction mixture containing 3aa and 4aa
The solvent was removed under reduced pressure (10–20 Torr). The reaction residue was diluted with a mixture of solvents PE/CHCl3/EA in a volume ratio of 1/2/2 (50 mL) and then filtered from the precipitate using SiO2 (d = 20 mm, h = 80 mm). The precipitate was washed with a mixture of solvents PE/CHCl3/EA in a volume ratio of 1/2/2 (3 × 30 mL). The combined organic phases were concentrated under reduced pressure (10–20 Torr). The yields of 3aa and 4aa were determined by 1H NMR using 1,4-dinitrobenzene as an internal standard.General procedure 2. Synthesis of β-hydroxysulfones 3aa–3ab and β-ketosulfones 4aa–4ab (Table 2)
To a solution of styrene 1a–1e (300 mg, 1.87–2.88 mmol) in 25 mL of CH3CN–H2O (5:1), CuBr (3.74–5.76 mmol, molar ratio 2 mol mol−11a–1e) and sulfonylhydrazide 2a–2b (1.87–2.88 mmol, molar ratio 1 mol 2/mol 1a–1e) were added. The mixture was stirred under an oxygen atmosphere for 7 h at 40 °C, then for 12 h at room temperature. Then, the reaction mixture was treated as described above (General procedure 1). The yields of 3aa, 3ba, 3ca, 3da, 3ea, 3ab and 4aa, 4ba, 4ca, 4da, 4ea, 4ab were determined by 1H NMR using 1,4-dinitrobenzene as an internal standard. The products 3aa, 3ba, 3ca, 3da, 3ea, 3ab and 4aa, 4ba, 4ca, 4da, 4ea, 4ab were isolated by chromatography on SiO2 with elution using PE–EA in a linear gradient of the latter from 10 to 40 vol%.
General procedure 3. Synthesis of β-hydroxysulfones 3aa–3ab using NaBH4 (Table 2, yield in parentheses)
The reaction mixture was treated as described above (General procedure 1). Then, the residue was diluted with 10 mL of THF-MeOH (1:1) mixture and NaBH4 (molar ratio 3 mol mol−14aa, 4ba, 4ca, 4da, 4ea, 4ab) was added with vigorous stirring. The mixture was stirred for 3 h at 0–5 °C. The solvent was removed under reduced pressure (10–20 Torr). The residue was diluted with EA (50 mL) and washed with water (2 × 5 mL), brine (3 × 5 mL) and again water (2 × 5 mL), dried over Na2SO4 and concentrated under reduced pressure (10–20 Torr). The desired products 3aa, 3ba, 3ca, 3da, 3ea, 3ab were isolated by chromatography on SiO2 with elution using PE–EA in a linear gradient of the latter from 10 to 40 vol%.
:EA, 5:1). 1H NMR (CDCl3), δ: 2.45 (s, 3H, CH3), 3.31 (dd, J = 14.3, 1.8 Hz, 1H), 3.47 (dd, J = 10.0, 14.3 Hz, 1H), 3.76 (d, J = 2.0 Hz, 1H), 5.23 (ddd, J = 10.3, 2.0, 1.8 Hz, 1H), 7.23–7.32 (m, 5H), 7.37 (d, J = 8.1 Hz, 2H), 7.82 (d, J = 8.1 Hz, 2H). 13C NMR (CDCl3), δ: 21.6, 63.9, 68.4, 125.6, 127.9, 128.2, 128.6, 130.0, 136.1, 140.7, 145.1. Calculated for C15H16O3S C: 65.19%, H: 5.84%, S: 11.60%. Found C: 65.12%, H: 5.78%, S: 11.66%. HRMS (ESI) m/z [M + Na]+: calculated for [C15H16NaO3S]+: 299.0718. Found: 299.0712. IR (KBr), ν, cm−1: 3496, 1391, 1286, 1167, 1137, 1087, 1064, 1020, 998, 834, 818, 779, 747, 706, 640, 555, 537, 514, 500, 462.
:EA, 3:1). 1H NMR (CDCl3), δ: 2.08 (s, 3H), 2.46 (s, 3H), 3.22 (dd, J = 14.5, 1.3 Hz, 1H), 3.39 (dd, J = 14.5, 9.8 Hz, 1H), 3.69 (s, 1H), 5.42 (d, J = 9.8 Hz, 1H), 7.07 (dd, J = 7.2, 2.0 Hz, 1H), 7, 11–7.24 (m, 2H), 7.38 (d, J = 8.1 Hz, 2H), 7.48 (dd, J = 7.2, 1.7 Hz, 1H), 7.85 (d, J = 8.1 Hz, 2H). 13C NMR (CDCl3), δ: 18.5, 21.6, 62.9, 65.0, 125.2, 126.5, 127.9, 128.0, 130.0, 130.5, 133.6, 136.0, 138.7, 145.2. Calculated for C16H18O3S C: 66.18%, H: 6.25%, S: 11.04%. Found C: 66.15%, H: 6.21%, S: 10.89%. HRMS (ESI) m/z [M + Na]+: calculated for [C16H18NaO3S]+: 313.0874. Found: 313.0869. IR (KBr), ν, cm−1: 3517, 1299, 1287, 1247, 1236, 1199, 1189, 1170, 1158, 1142, 1086, 1047, 857, 803, 758, 749, 721, 638, 564, 518, 506, 456.
:EA, 3:1). 1H NMR (DMSO-d6), δ: 1.16 (d, J = 6.9 Hz, 6H), 2.39 (s, 3H), 2.83 (m, J = 6.9 Hz, 1H), 3.50 (dd, J = 14.5, 3.7 Hz, 1H), 3.67 (dd, J = 14.5, 8.5 Hz, 1H), 4.96 (ddd, J = 8.5, 4.7, 3.7 Hz, 1H), 5.52 (d, J = 4.7 Hz, 1H), 7.13 (d, J = 8.2 Hz, 2H), 7.20 (d, J = 8.2 Hz, 2H), 7.38 (d, J = 8.2 Hz, 2H), 7.75 (d, J = 8.2 Hz, 2H). 13C NMR (DMSO-d6), δ: 21.0, 23.9, 33.1, 63.0, 67.8, 126.0, 126.1, 127.8, 129.4, 137.6, 140.5, 143.7, 147.6. Calculated for C18H22O3S C: 67.89%, H: 6.96%, S: 10.07%. Found C: 67.87%, H: 7.01%, S: 10.14%. HRMS (ESI) m/z [M + Na]+: calculated for [C18H22NaO3S]+: 341.1187. Found: 341.1178. IR (KBr), ν, cm−1: 3500, 2966, 1410, 1302, 1287, 1253, 1170, 1140, 1086, 1052, 1001, 862, 851, 822, 776, 734, 635, 597, 568, 547, 532, 509, 471, 449.
:EA, 3:1). 1H NMR (DMSO-d6), δ: 1.24 (s, 9H), 2.39 (s, 3H), 3.50 (dd, J = 14.6, 3.8 Hz, 1H), 3.67 (dd, J = 14.6, 8.5 Hz, 1H), 4.96 (ddd, J = 8.5, 4.7, 3.8 Hz, 1H), 5.52 (d, J = 4.7 Hz, 1H), 7.20 (d, J = 8.2 Hz, 2H), 7.28 (d, J = 8.2 Hz, 2H), 7.37 (d, J = 8.1 Hz, 2H), 7.75 (d, J = 8.1 Hz, 2H). 13C NMR (DMSO-d6), δ: 21.0, 31.1, 34.1, 62.9, 67.7, 124.8, 125.8, 127.7, 129.4, 137.6, 140.0, 143.6, 149.8. Calculated for C19H24O3S C: 68.64%, H: 7.28%, S: 9.64%. Found C: 68.57%, H: 6.94%, S: 9.51%. HRMS (ESI) m/z [M + Na]+: calculated for [C19H24NaO3S]+: 313.0874. Found: 313.0869. IR (KBr), ν, cm−1: 3521, 2962, 1303, 1289, 1242, 1174, 1140, 1113, 1087, 1057, 864, 844, 823, 773, 737, 636, 579, 543, 528, 506.
:EA, 3:1). 1H NMR (DMSO-d6), δ: 2.40 (s, 3H), 3.55 (dd, J = 14.6, 4.0 Hz, 1H), 3.67 (dd, J = 14.6, 8.2 Hz, 1H), 4.97 (ddd, J = 8.2, 5.0, 4.0 Hz, 1H), 5.70 (d, J = 5.0 Hz, 1H), 7.32 (s, 4H), 7.39 (d, J = 8.1 Hz, 1H), 7.75 (d, J = 8.1 Hz, 1H). 13C NMR (DMSO-d6), δ: 21.0, 62.6, 67.3, 127.8, 128.0, 128.1, 129.4, 131.9, 137.5, 141.9, 143.8. Calculated for C15H15ClO3S C: 57.97%, H: 4.86%, Cl: 11.41%, S: 10.32%. Found C: 57.95%, H: 4.93%, Cl: 11.34%, S: 10.25%. HRMS (ESI) m/z [M + Na]+: calculated for: [C15H15ClNaO3S]+: 333.0328. Found: 333.0323. IR (KBr), ν, cm−1: 3485, 1311, 1300, 1287, 1160, 1145, 1138, 1087, 1076, 1064, 1013, 813, 714, 562, 511, 502.
:EA, 5:1). 1H NMR (DMSO-d6), δ: 3.55 (dd, J = 14.6, 3.3 Hz, 1H), 3.76 (dd, J = 14.6, 9.2 Hz, 1H), 5.00 (ddd, J = 9.2, 4.8, 3.3 Hz, 1H), 5.62 (d, J = 4.8 Hz, 1H), 7.19–7.34 (m, 5H), 7.65 (d, J = 8.4 Hz, 2H), 7.99 (d, J = 8.4 Hz, 2H). 13C NMR (DMSO-d6), δ: 62.6, 67.9, 101.9, 126.1, 127.4, 128.2, 129.5, 137.8, 140.3, 142.9. Calculated for C14H13IO3S C: 43.31%, H: 3.38%, I: 32.69%, S: 8.26%. Found C: 43.28%, H: 3.31%, I: 32.32%, S: 8.09%. HRMS (ESI) m/z [M + Na]+: calculated for [C14H13INaO3S]+: 410.9528. Found: 410.9522. IR (KBr), ν, cm−1: 3464, 1384, 1303, 1270, 1135, 1083, 1061, 1003, 993, 817, 745, 701, 566, 549, 531.
:EA, 2:1). 1H NMR (CDCl3), δ: 2.43 (s, 3H), 4.73 (s, 2H), 7.32 (d, J = 8.2 Hz, 2H), 7.46 (dd, J = 7.5, 7.3 Hz, 2H), 7.61 (t, J = 7.5 Hz, 1H), 7.77 (d, J = 8.2 Hz, 2H), 7.94 (d, J = 7.3 Hz, 2H). 13C NMR (CDCl3), δ: 21.6, 63.5, 128.5, 128.7, 129.2, 129.7, 134.2, 135.7, 135.8, 145.2, 188.1. Calculated for C15H14O3S C: 65.67%, H: 5.14%, S: 11.69%. Found C: 65.57%, H: 5.34%, S: 11.59%. HRMS (ESI) m/z [M + Na]+: calculated for [C15H14NaO3S]+: 297.0561. Found: 297.0556. IR (KBr), ν, cm−1: 1680, 1596, 1320, 1271, 1150, 1087, 993, 750, 739, 686, 590, 535, 503.
:EA, 3:1). 1H NMR (CDCl3), δ: 2.45 (s, 6H), 4.70 (s, 2H), 7.23–7.31 (m, 2H), 7.31–7.36 (m, 2H), 7.39–7.46 (m, 1H), 7.71–7.79 (m, 3H). 13C NMR (CDCl3), δ: 21.4, 21.6, 65.5, 125.8, 128.4, 129.7, 130.3, 132.2, 132.7, 135.7, 136.0, 139.9, 145.1, 190.5. Calculated for C16H16O3S C: 66.64%, H: 5.59%, S: 11.12%. Found C: 66.38%, H: 5.80%, S: 11.29%. HRMS (ESI) m/z [M + Na]+: calculated for [C16H16NaO3S]+: 311.0718. Found: 311.0714. IR (KBr), ν, cm−1: 2956, 2910, 1685, 1314, 1291, 1142, 1084, 978, 823, 761, 747, 556, 517.
:EA, 3:1). 1H NMR (CDCl3), δ: 1.26 (d, J = 7.0 Hz, 6H), 2.43 (s, 3H), 2.97 (m, J = 7.0 Hz, 1H), 4.69 (s, 2H), 7.32 (2d, J = 8.2, 8.4 Hz, 4H), 7.76 (d, J = 8.2 Hz, 2H), 7.87 (d, J = 8.4 Hz, 2H). 13C NMR (CDCl3), δ: 21.6, 23.5, 34.3, 63.5, 126.9, 128.6, 129.6, 129.7, 133.7, 135.8, 145.2, 156.1, 187.6. Calculated for C18H20O3S C: 68.33%, H: 6.37%, S: 10.13%. Found C: 67.86%, H: 6.82%, S: 9.70%. HRMS (ESI) m/z [M + Na]+: calculated for [C18H20NaO3S]+: 339.1031. Found: 339.1025. IR (KBr), ν, cm−1: 2953, 1686, 1311, 1290, 1183, 1142, 1084, 828, 549.
:EA, 3:1). 1H NMR (CDCl3), δ: 1.34 (s, 9H), 2.44 (s, 3H), 4.69 (s, 2H), 7.32 (d, J = 7.9 Hz, 2H), 7.48 (d, J = 8.2 Hz, 2H), 7.76 (d, J = 7.9 Hz, 2H), 7.88 (d, J = 8.2 Hz, 2H). 13C NMR (CDCl3), δ: 21.7, 31.0, 35.2, 63.6, 125.8, 128.6, 129.3, 129.8, 133.3, 135.9, 145.2, 158.3, 187.6. Calculated for C19H22O3S C: 69.06%, H: 6.71%, S: 9.70%. Found C: 69.01%, H: 6.65%, S: 9.59%. HRMS (ESI) m/z [M + Na]+: calculated for [C19H22NaO3S]+: 353.1187. Found: 353.1182. IR (KBr), ν, cm−1: 1681, 1314, 1290, 1141, 1083, 828, 768, 590, 549, 516.
:EA, 3:1). 1H NMR (CDCl3), δ: 2.45 (s, 3H), 4.68 (s, 2H), 7.34 (d, J = 8.2 Hz, 2H), 7.45 (d, J = 8.6 Hz, 2H), 7.74 (d, J = 8.2 Hz, 2H), 7.90 (d, J = 8.6 Hz, 2H). 13C NMR (CDCl3), δ: 21.7, 63.7, 128.5, 129.2, 129.9, 130.7, 134.1, 135.6, 141.0, 145.5, 187.0. Calculated for C15H13ClO3S C: 58.35%, H: 4.24%, Cl: 11.48%, S: 10.38%. Found C: 58.37%, H: 4.31%, Cl: 10.98%, S: 9.93%. HRMS (ESI) m/z [M + Na]+: calculated for [C15H13ClNaO3S]+: 331.0172. Found: 331.0166. IR (KBr), ν, cm−1: 1679, 1589, 1315, 1290, 1277, 1148, 1091, 1083, 1004, 784, 759, 724, 537, 507.
:EA, 5:1). 1H NMR (CDCl3), δ: 4.73 (s, 2H), 7.49 (t, J = 7.2 Hz, 2H), 7.56–7.68 (m, 3H), 7.87–7.95 (m, 4H). 13C NMR (CDCl3), δ: 63.2, 102.5, 128.9, 129.2, 129.9, 134.5, 135.5, 138.3, 138.4, 187.8. Calculated for C14H11IO3S C: 43.54%, H: 2.87%, I: 32.86%, S: 8.30%. Found C: 43.58%, H: 3.01%, I: 32.65%, S: 8.25%. HRMS (ESI) m/z [M + Na]+: calculated for [C14H11INaO3S]+: 408.9371. Found: 408.9366. IR (KBr), ν, cm−1: 1677, 1563, 1379, 1314, 1273, 1151, 1002, 756, 741, 727, 565, 516.
General procedure 4. Synthesis of β-hydroxysulfones 3fa–3fe (Table 3)
To a solution of styrene 1f–1j (300 mg, 1.52–2.54 mmol) in 25 mL of CH3CN–H2O (5:1), CuBr (3.04–5.08 mmol, molar ratio 2 mol mol−11f–1j) and sulfonylhydrazide 2a–2e (1.52–2.54 mmol, molar ratio 1 mol 2/mol 1f–1j) were added. The mixture was stirred under an oxygen atmosphere for 7 h at 40 °C, then for 12 h at room temperature. Then, the reaction mixture was treated as described above (General procedure 1). The desired products 3fa, 3ga, 3ha, 3ia, 3ja, 3fc, 3fd, 3fe were isolated by chromatography on SiO2 with elution using PE–EA in a linear gradient of the latter from 10 to 40 vol%.
:EA, 2:1). 1H NMR (CDCl3), δ: 1.71 (d, J = 1.1 Hz, 3H), 2.39 (s, 3H), 3.61 (dd, J = 14.6, 1.1 Hz, 1H), 3.72 (d, J = 14.6, 1.1 Hz, 1H), 4.66 (br s, 1H), 7.14–7.24 (m, 5H), 7.26–7.34 (m, 2H), 7.49 (dd, J = 8.2, 1.0 Hz, 2H). 13C NMR (CDCl3), δ: 21.5, 30.7, 66.6, 73.0, 124.5, 127.0, 127.4, 128.1, 129.6, 137.3, 144.4. Calculated for C16H18O3S C: 66.18%, H: 6.25%, S: 11.04%. Found C: 66.23%, H: 6.06%, S: 11.12%. HRMS (ESI) m/z [M + Na]+: calculated for [C16H18NaO3S]+: 313.0874. Found: 313.0879. IR (KBr), ν, cm−1: 3500, 2973, 1451, 1355, 1300, 1268, 1247, 1182, 1155, 1119, 1081, 1036, 1024, 1017, 947, 858, 813, 767, 707, 637, 570, 555, 532, 509, 477.
:EA, 2:1). 1H NMR (CDCl3), δ: 1.62 (s, 3H), 2.39 (s, 3H), 3.55 (d, J = 14.8 Hz, 1H), 3.69 (d, J = 14.8 Hz, 1H), 7.07 (d, J = 8.7 Hz, 2H) 7.15 (d, J = 8.7 Hz, 2H) 7.15 (d, J = 8.3 Hz, 2H), 7.41 (d, J = 8.3 Hz, 2H). 13C NMR (CDCl3), δ: 21.5, 30.9, 66.3, 72.7, 126.2, 127.5, 128.2, 129.7, 133.1, 137.0, 142.8, 144.7. Calculated for C16H17ClO3S C: 59.16%, H: 5.28%, Cl: 10.91%, S: 9.87%. Found C: 58.99%, H: 5.29%, Cl: 11.04%, S: 9.98%. HRMS (ESI) m/z [M + Na]+: calculated for [C16H17ClNaO3S]+: 347.0485. Found: 347.0479. IR (KBr), ν, cm−1: 3496, 1308, 1302, 1252, 1158, 1128, 1082, 1044, 849, 771, 645, 543, 522, 460.
:EA, 3:1).1H NMR (DMSO-d6), δ: 1.55 (s, 3H), 2.37 (s, 3H), 3.77 (d, J = 14.8 Hz, 1H), 3.84 (d, J = 14.8 Hz, 1H), 7.24–7.35 (m, 6H), 7.52 (d, J = 8.0 Hz, 2H). 13C NMR (DMSO-d6), δ: 21.0, 30.0, 66.0, 71.5, 119.8, 127.5, 127.6, 129.2, 130.3, 138.2, 143.4, 145.4. Calculated for C16H17BrO3S C: 52.04%, H: 4.64%, Br: 21.64%, S: 8.68%. Found C: 52.14%, H: 4.73%, Br: 21.12%, S: 8.47%. HRMS (ESI) m/z [M + Na]+: calculated for [C16H17BrNaO3S]+: 390.9979. Found: 390.9974. IR (KBr), ν, cm−1: 3489, 1251, 1158, 1127, 1081, 771, 639, 585, 540, 522, 486.
:EA, 2:1). 1H NMR (DMSO-d6), δ: 1.57 (s, 3H), 2.33 (s, 3H), 3.84 (d, J = 14.8 Hz, 1H), 4.01 (d, J = 14.8 Hz, 1H), 5.71 (s, 1H), 7.25 (d, J = 8.0 Hz, 2H), 7.51 (d, J = 8.0 Hz, 2H), 7.64 (d, J = 8.9 Hz, 2H), 8.02 (d, J = 8.8 Hz, 2H). 13C NMR (DMSO-d6), δ: 21.0, 30.5, 65.7, 71.8, 122.6, 126.7, 127.7, 129.2, 138.1, 143.6, 146.1, 153.7. Calculated for C16H17NO5S C: 57.30%, H: 5.11%, N: 4.18%, S: 9.56%. Found C: 57.28%, H: 5.08%, N: 4.16%, S: 9.48%. HRMS (ESI) m/z [M + Na]+: calculated for [C16H17NNaO5S]+: 358.0725. Found: 358.0713. IR (KBr), ν, cm−1: 3480, 1520, 1349, 1310, 1301, 1291, 1268, 1147, 1121, 1084, 855, 815, 757, 537, 518.
:EA, 3:1). 1H NMR (CDCl3), δ: 1.66 (s, 3H), 2.37 (s, 3H), 3.54 (d, J = 14.5 Hz, 1H), 3.67 (d, J = 14.5 Hz 1H), 3.74 (s, 3H), 6.68 (d, J = 8.8 Hz, 2H), 7.16 (d, J = 8.3 Hz, 2H), 7.17 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 8.3 Hz, 2H). 13C NMR (CDCl3), δ: 21.5, 30.7, 55.2, 66.8, 72.8, 113.5, 125.8, 127.5, 129.6, 136.6, 137.4, 144.3, 158.7. Calculated for C17H20O4S C: 63.73%, H: 6.29%, S: 10.01%. Found C: 63.88%, H: 6.29%, S: 10.01%. HRMS (ESI) m/z [M + Na]+: calculated for [C17H20NaO4S]+: 343.0980. Found: 343.0975. IR (KBr), ν, cm−1: 3472, 1607, 1598, 1514, 1309, 1292, 1251, 1183, 1146, 1120, 1083, 1032, 832, 823, 810, 766, 676, 557, 536, 513.
:EA, 3:1). 1H NMR (DMSO-d6), δ: 1.60 (s, 3H), 3.86 (s, 2H), 4.75 (br s, 1H), 7.11–7.23 (m, 3H), 7.33–7.39 (m, 2H), 7.62 (d, J = 8.6 Hz, 2H), 7.70 (d, J = 8.6 Hz, 2H). 13C NMR (DMSO-d6), δ: 29.9, 66.1, 71.7, 125.0, 126.4, 127.1, 127.6, 129.8, 131.7, 140.6, 146.5. Calculated for C15H15BrO3S C: 50.71%, H: 4.26%, Br: 22.49%, S: 9.03%. Found C: 50.78%, H: 4.31%, Br: 22.48%, S: 9.02%. HRMS (ESI) m/z [M + Na]+: calculated for [C15H15BrNaO3S]+: 376.9823. Found: 376.9821. IR (KBr), ν, cm−1: 3507, 1576, 1392, 1312, 1295, 1270, 1149, 1122, 1084, 1067, 1010, 942, 821, 775, 765, 714, 701, 579, 545, 528, 411.
:EA, 2:1). 1H NMR (CDCl3), δ: 1.68 (s, 3H), 3.58 (d, J = 14.7 Hz, 1H), 3.70 (d, J = 14.7 Hz, 1H), 3.82 (s, 3H), 6.81 (d, J = 8.9 Hz, 2H), 7.11–7.24 (m, 3H), 7.25–7.31 (m, 2H), 7.50 (d, J = 8.9 Hz, 2H). 13C NMR (CDCl3), δ: 30.8, 55.6, 66.7, 73.0, 114.2, 124.6, 127.1, 128.2, 129.7, 131.8, 144.5, 163.5. Calculated for C16H18O4S C: 62.72%, H: 5.92%, S: 10.47%. Found C: 62.81%, H: 5.95%, S: 10.46%. HRMS (ESI) m/z [M + Na]+: calculated for [C16H18NaO4S]+: 329.0824. Found: 329.0813. IR (KBr), ν, cm−1: 3501, 1594, 1497, 1307, 1295, 1261, 1249, 1151, 1117, 1079, 1026, 830, 758, 697, 570, 529, 480, 468.
:EA, 3:1). 1H NMR (DMSO-d6), δ: 1.61 (s, 3H), 3.93–4.05 (m, 2H), 5.44 (s, 1H), 7.08–7.23 (m, 3H), 7.31–7.39 (m, 2H), 7.96 (d, J = 8.7 Hz, 2H), 8.29 (d, J = 8.7 Hz, 2H). 13C NMR (DMSO-d6), δ: 29.9, 65.8, 71.6, 123.7, 124.9, 126.4, 127.5, 129.4, 146.3, 146.6, 149.7. Calculated for C15H15NO5S C: 56.06%, H: 4.70%, N: 4.36%, S: 9.98%. Found C: 56.04%, H: 4.85%, N: 4.21%, S: 9.96%. HRMS (ESI) m/z [M + Na]+: calculated for [C15H15NNaO5S]+: 344.0569. Found: 344.0563. IR (KBr), ν, cm−1: 3492, 1525, 1350, 1305, 1148, 1121, 1083, 849, 771, 742, 579, 525.
Acknowledgements
This work was supported by the Russian Science Foundation (Grant 14-23-00150).References
- H. Eto, Y. Kaneko, S. Takeda, M. Tokizawa, S. Sato, K. Yoshida, S. Namiki, M. Ogawa, K. Maebashi, K. Ishida, M. Matsumoto and T. Asaoka, Chem. Pharm. Bull., 2001, 49, 173 CrossRef CAS PubMed.
- B. J. A. Furr, Horm. Res., 1989, 32, 69 CrossRef CAS PubMed.
- T. Sato, Y. Okumura, J. Itai and T. Fujisawa, Chem. Lett., 1988, 17, 1537 CrossRef.
- M. Julia and J.-M. Paris, Tetrahedron Lett., 1973, 14, 4833 CrossRef.
- N. Suryakiran, T. Srikanth Reddy and Y. Venkateswarlu, J. Sulfur Chem., 2007, 28, 513 CrossRef CAS.
- A. K. Maiti and P. Bhattacharyya, Tetrahedron, 1994, 50, 10483 CrossRef CAS.
- D. J. Berrisford, P. A. Lovell, N. R. Suliman and A. Whiting, Chem. Commun., 2005, 5904 RSC.
- S. Narayana Murthy, B. Madhav, V. Prakash Reddy, K. Rama Rao and Y. V. D. Nageswar, Tetrahedron Lett., 2009, 50, 5009 CrossRef CAS.
- M. C. Bernabeu, P. Bonete, F. Caturla, R. Chinchilla and C. Nájera, Tetrahedron: Asymmetry, 1996, 7, 2475 CrossRef CAS.
- P. Bertus, P. Phansavath, V. Ratovelomanana-Vidal, J. P. Genêt, A. R. Touati, T. Homri and B. B. Hassine, Tetrahedron Lett., 1999, 40, 3175 CrossRef CAS.
- A. L. Moure, R. G. Arrayas and J. C. Carretero, Chem. Commun., 2011, 47, 6701 RSC.
- W. Autenrieth, Liebigs Ann. Chem., 1890, 259, 362 CrossRef.
- W. E. Truce and A. M. Murphy, Chem. Rev., 1951, 48, 69 CrossRef CAS PubMed.
- W. Wei, J. Wen, D. Yang, H. Jing, J. You and H. Wang, RSC Adv., 2015, 5, 4416 RSC.
- Q. Lu, J. Zhang, F. Wei, Y. Qi, H. Wang, Z. Liu and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 7156 CrossRef CAS PubMed.
- T. Taniguchi, A. Idota and H. Ishibashi, Org. Biomol. Chem., 2011, 9, 3151 CAS.
- X. Tang, L. Huang, Y. Xu, J. Yang, W. Wu and H. Jiang, Angew. Chem., Int. Ed., 2014, 53, 4205 CrossRef CAS PubMed.
- W. Wei, J. Li, D. Yang, J. Wen, Y. Jiao, J. You and H. Wang, Org. Biomol. Chem., 2014, 12, 1861 CAS.
- V. Nair, A. Augustine, T. G. George and L. G. Nair, Tetrahedron Lett., 2001, 42, 6763 CrossRef CAS.
- Y. Gao, W. Wu, Y. Huang, K. Huang and H. Jiang, Org. Chem. Front., 2014, 1, 361 RSC.
- W. Wei, J. Wen, D. Yang, J. Du, J. You and H. Wang, Green Chem., 2014, 16, 2988 RSC.
- L. Zhang, S. Chen, Y. Gao, P. Zhang, Y. Wu, G. Tang and Y. Zhao, Org. Lett., 2016, 18, 1286 CrossRef CAS PubMed.
- F. Xiao, H. Chen, H. Xie, S. Chen, L. Yang and G. J. Deng, Org. Lett., 2014, 16, 50 CrossRef CAS PubMed.
- J. K. Qiu, W. J. Hao, D. C. Wang, P. Wei, J. Sun, B. Jiang and S. J. Tu, Chem. Commun., 2014, 50, 14782 RSC.
- W. Wei, J. Wen, D. Yang, M. Guo, Y. Wang, J. You and H. Wang, Chem. Commun., 2015, 51, 768 RSC.
- K. M. Gligorich and M. S. Sigman, Chem. Commun., 2009, 3854 RSC.
- Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381 RSC.
- A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851 CrossRef CAS PubMed.
- W. Wei, J. Wen, D. Yang, M. Wu, J. You and H. Wang, Org. Biomol. Chem., 2014, 12, 7678 CAS.
- L. C. Anderson and N. W. MacNaughton, J. Am. Chem. Soc., 1942, 64, 1456 CrossRef CAS.
- R. Barnes, J. Graham and M. Taylor, J. Org. Chem., 1958, 23, 1561 CrossRef.
- S. Chakraborty, P. Bhattacharya, H. Dai and H. Guan, Acc. Chem. Res., 2015, 48, 1995 CrossRef CAS PubMed.
- X. Li, X. Xu and C. Zhou, Chem. Commun., 2012, 48, 12240 RSC.
- W. Wei, C. Liu, D. Yang, J. Wen, J. You, Y. Suo and H. Wang, Chem. Commun., 2013, 49, 10239 RSC.
- J. Wen, W. Wei, S. Xue, D. Yang, Y. Lou, C. Gao and H. Wang, J. Org. Chem., 2015, 80, 4966 CrossRef CAS PubMed.
- V. K. Sharma and F. J. Millero, Environ. Sci. Technol., 1988, 22, 768 CrossRef CAS PubMed.
- B. M. Voelker, D. L. Sedlak and O. C. Zafiriou, Environ. Sci. Technol., 2000, 34, 1036 CrossRef CAS.
- M. González-Dávila, J. M. Santana-Casiano, A. G. González, N. Pérez and F. J. Millero, Mar. Chem., 2009, 115, 118 CrossRef.
- X. Yuan, A. N. Pham, G. Xing, A. L. Rose and T. D. Waite, Environ. Sci. Technol., 2012, 46, 1527 CrossRef CAS PubMed.
- P. Zhou, J. Zhang, Y. Zhang, Y. Liu, J. Liang, B. Liu and W. Zhang, RSC Adv., 2016, 6, 38541 RSC.
- G. W. Filson and J. H. Walton, J. Phys. Chem., 1931, 36, 740 CrossRef.
- P. M. Henry, Inorg. Chem., 1966, 5, 688 CrossRef CAS.
- A. S. Jhaveri and M. M. Sharma, Chem. Eng. Sci., 1967, 22, 1 CrossRef CAS.
- R. D. Gray, J. Am. Chem. Soc., 1969, 91, 56 CrossRef CAS.
- T. Taniguchi, H. Zaimoku and H. Ishibashi, Chem.–Eur. J., 2011, 17, 4307 CrossRef CAS PubMed.
- M. Kharasch and A. Fono, J. Org. Chem., 1958, 23, 324 Search PubMed.
- M. S. Kharasch and A. Fono, J. Org. Chem., 1959, 24, 606 CrossRef CAS.
- M. S. Kharasch, G. Sosnovsky and N. C. Yang, J. Am. Chem. Soc., 1959, 81, 5819 CrossRef CAS.
- G. Sosnovsky and N. C. Yang, J. Org. Chem., 1960, 25, 899 CrossRef CAS.
- J. K. Kochi, J. Am. Chem. Soc., 1961, 83, 3162 CrossRef CAS.
- J. K. Kochi, J. Am. Chem. Soc., 1963, 85, 1958 CrossRef CAS.
- S. H. Oh, Y. R. Malpani, N. Ha, Y. S. Jung and S. B. Han, Org. Lett., 2014, 16, 1310 CrossRef CAS PubMed.
- K. Sun, Y. Lv, Z. Zhu, Y. Jiang, J. Qi, H. Wu, Z. Zhang, G. Zhang and X. Wang, RSC Adv., 2015, 5, 50701 RSC.
- S. F. Zhou, D. P. Li, K. Liu, J. P. Zou and O. T. Asekun, J. Org. Chem., 2015, 80, 1214 CrossRef CAS PubMed.
- H. R. Lucas, L. Li, A. A. Sarjeant, M. A. Vance, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2009, 131, 3230–3245 CrossRef CAS PubMed.
- S. Lim, M. Ji, X. Wang, C. Lee and H.-Y. Jang, Eur. J. Org. Chem., 2015, 591 CrossRef CAS.
- M. Catir, H. Kilic, V. Nardello-Rataj, J. M. Aubry and C. Kazaz, J. Org. Chem., 2009, 74, 4560 CrossRef CAS PubMed.
- M. Nakano, K. Takayama, Y. Shimizu, Y. Tsuji, H. Inaba and T. Migita, J. Am. Chem. Soc., 1976, 98, 1974 CrossRef CAS.
- A. M. Tsedilin, A. N. Fakhrutdinov, D. B. Eremin, S. S. Zalesskiy, A. O. Chizhov, N. G. Kolotyrkina and V. P. Ananikov, Mendeleev Commun., 2015, 25, 454–456 CrossRef CAS.
- A. J. Perkowski, W. You and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 7580 CrossRef CAS PubMed.
- G. L. Backes, B. S. Jursic and D. M. Neumann, Bioorg. Med. Chem., 2015, 23, 3397 CrossRef CAS PubMed.
- G. Zhang, X. Xie, Y. Wang, X. Wen, Y. Zhao and C. Ding, Org. Biomol. Chem., 2013, 11, 2947 CAS.
- C. B. Tripathi and S. Mukherjee, Angew. Chem., Int. Ed., 2013, 52, 8450 CrossRef CAS PubMed.
- T. N. Gieshoff, M. Villa, A. Welther, M. Plois, U. Chakraborty, R. Wolf and A. Jacobi von Wangelin, Green Chem., 2015, 17, 1408 RSC.
- H. Lebel, M. Davi, S. Diez-Gonzalez and S. P. Nolan, J. Org. Chem., 2007, 72, 144 CrossRef CAS PubMed.
- N. C. Deno, F. A. Kish and H. J. Peterson, J. Am. Chem. Soc., 1965, 87, 2157 CrossRef CAS.
- H. Zinner, H. Brenken, W. Braun, I. Falk, E. Fechtner and E. Hähner, Liebigs Ann. Chem., 1959, 622, 133 CrossRef CAS.
- C. B. Tripathi and S. Mukherjee, Org. Lett., 2014, 16, 3368 CrossRef CAS PubMed.
- K. M. Khan, U. Salar, M. I. Fakhri, M. Taha, A. Hameed, S. Perveen and W. Voelter, Lett. Org. Chem., 2015, 12, 637 CrossRef CAS.
- N. Taniguchi, J. Org. Chem., 2015, 80, 7797 CrossRef CAS PubMed.
- Y. Tang, Y. Zhang, K. Wang, X. Li, X. Xu and X. Du, Org. Biomol. Chem., 2015, 13, 7084 CAS.
- S. Handa, J. C. Fennewald and B. H. Lipshutz, Angew. Chem., Int. Ed., 2014, 53, 3432 CrossRef CAS PubMed.
- D. F. Tavares and P. F. Vogt, Can. J. Chem., 1967, 45, 1519 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of all synthesized compounds. See DOI: 10.1039/c6ra19190h |
| This journal is © The Royal Society of Chemistry 2016 |