
Manganese oxide films with controlled oxidation state for water splitting devices through a combination of atomic layer deposition and post-deposition annealing†
 *a
*a
Abstract
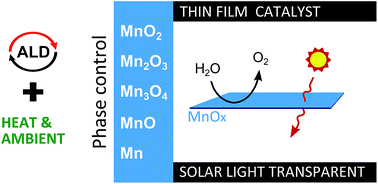
Solar hydrogen devices combine the power of photovoltaics and water electrolysis to produce hydrogen in a hybrid form of energy production. To engineer these into integrated devices (i.e. a water splitting catalyst on top of a PV element), the need exists for thin film catalysts that are both transparent for solar light and efficient in water splitting. Manganese oxides have already been shown to exhibit good water splitting performance, which can be further enhanced by conformal coating on high surface-area structures. The latter can be achieved by atomic layer deposition (ALD). However, to optimize the catalytic and transparency properties of the water splitting layer, an excellent control over the oxidation state of the manganese in the film is required. So far MnO, Mn3O4 and MnO2 ALD have been shown, while Mn2O3 is the most promising catalyst. Therefore, we investigated the post-deposition oxidation and reduction of MnO and MnO2 ALD films, and derived strategies to achieve every phase in the MnO–MnO2 range by tuning the ALD process and post-ALD annealing conditions. Thin film Mn2O3 is obtained by thermal reduction of ALD MnO2, without the need for oxidative high temperature treatments. The obtained Mn2O3 is examined for solar water splitting devices, and compared to the as-deposited MnO2. Both thin films show oxygen evolution activity and good solar light transmission.
 Please wait while we load your content...
Please wait while we load your content...

