
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMangifera indica, Ficus religiosa and Polyalthia longifolia leaf extract-assisted green synthesis of graphene for transparent highly conductive film†
Pankaj
Chamoli
a,
Raghunandan
Sharma
b,
Malay K.
Das
b and
Kamal K.
Kar
*ab
aAdvanced Nanoengineering Materials Laboratory, Materials Science Programme, Indian Institute of Technology Kanpur, Kanpur – 208016, India
bAdvanced Nanoengineering Materials Laboratory, Department of Mechanical Engineering, Indian Institute of Technology Kanpur, Kanpur – 208016, India. E-mail: kamalkk@iitk.ac.in; Fax: +91-512-2597408; Tel: +91-512-2597687
First published on 28th September 2016
Abstract
A green approach to synthesize graphene nanosheets (Gns) by reduction of graphene oxide (GO) using Mangifera indica, Ficus religiosa or Polyalthia longifolia leaf extract as reducing agent has been demonstrated. Further, transparent conducting films (TCFs) have been fabricated by spray coating of the synthesized Gns and the optoelectrical properties of the TCFs have been investigated. Of the three leaf extracts, Mangifera indica offers the best alternative reducing agent for the green synthesis of Gns at large scale. Raman spectroscopy reveals that GO has been deoxygenated significantly, with a Raman D to G band intensity ratio of ∼1.21, while the attainment of a C/O ratio of ∼4.58 in the synthesized Gns has been confirmed by elemental analysis. The TCF fabricated after thermal graphitization (900 °C for 1 h) of the spray coated Gns film shows a sheet resistance of ∼2.08 kΩ □−1 and transmittance of ∼57.31% at 550 nm. The study suggests significant potential for the application of plant leaf extracts as non-toxic reducing agents in large-scale production of Gns. Apart from providing an alternative to hazardous reducing agents, this approach opens the possibility of preparation of Gns at large scale by using more accessible natural resources with mild synthesis conditions.
1. Introduction
Graphene, a flat monolayer of sp2 hybridized carbon arranged in a honeycomb lattice, has received great attention in the scientific community around the globe owing to its outstanding properties such as excellent room temperature carrier mobility ∼200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1, high optical transmittance ∼97.7%, fracture strength ∼130 GPa and excellent thermal conductivity ∼5000 W m−1 K−1; and it has opened broad areas of research in nanoscience and technology.1 Graphene has 2D planar structure and is a basic building block for all graphitic materials including fullerenes (0D), carbon nanotubes (1D), and graphite (3D), from nanoscale to bulk. The remarkable properties of graphene have potential application in modern electronic devices such as touch screens, displays, thin-film transistors, thin film photovoltaics and flexible electronics; and in various biomedical applications such as tissue engineering, drug delivery and bio-imaging.2–8 Several methods, including chemical vapor deposition (CVD), arc discharge, monolayer or bilayer extraction from exfoliated graphite, wet chemical route via chemical reduction of graphene oxide (GO), etc., have been employed for synthesis of graphene nanosheets (Gns).9–16 Among these, wet chemical routes are the most versatile and cost effective. In this process, Gns are produced by chemical reduction of GO in the presence of a reducing agent such as hydrazine and its derivatives, sodium borohydride,12 hydroquinone,13 hydroiodic acid/acetic acid,14,15 polyethyleneimine,16etc.
000 cm2 V−1 s−1, high optical transmittance ∼97.7%, fracture strength ∼130 GPa and excellent thermal conductivity ∼5000 W m−1 K−1; and it has opened broad areas of research in nanoscience and technology.1 Graphene has 2D planar structure and is a basic building block for all graphitic materials including fullerenes (0D), carbon nanotubes (1D), and graphite (3D), from nanoscale to bulk. The remarkable properties of graphene have potential application in modern electronic devices such as touch screens, displays, thin-film transistors, thin film photovoltaics and flexible electronics; and in various biomedical applications such as tissue engineering, drug delivery and bio-imaging.2–8 Several methods, including chemical vapor deposition (CVD), arc discharge, monolayer or bilayer extraction from exfoliated graphite, wet chemical route via chemical reduction of graphene oxide (GO), etc., have been employed for synthesis of graphene nanosheets (Gns).9–16 Among these, wet chemical routes are the most versatile and cost effective. In this process, Gns are produced by chemical reduction of GO in the presence of a reducing agent such as hydrazine and its derivatives, sodium borohydride,12 hydroquinone,13 hydroiodic acid/acetic acid,14,15 polyethyleneimine,16etc.
However, application of such toxic reducing agents for large-scale production is hazardous for human health as well as for the ecosystem. To minimize the environmental impacts, natural/non-toxic reducing agents such as melatonin, vitamin C, sugar, etc. have been investigated as reducing agents for GO reduction.17 In addition, plant extracts consisting of different biomolecules, including proteins, amino acids, enzymes, vitamins, etc., are being considered as easily accessible, cost efficient and environmentally friendly sources of reducing agents for reduction of GO.18–20 Especially, leaf extracts of Colocasia esculenta,18,19Mesua ferrea Linn.,18Tamarix gallica,20etc. have been explored for their ability to reduce GO. However, owing to the low concentration and poor activity of such plant extracts, the GO reduction process is time consuming and inefficient. Hence, exploring other plants with high concentrations of phytochemicals with reducing properties is of significant importance towards providing green alternatives for reduction of GO. Ficus religiosa, Mangifera indica, and Polyalthia longifolia leaves, which have had sacred importance in Hindu mythology from ancient times in India, exhibit several pharmacological activities owing to their anti-inflammatory, anti-oxidant, anti-allergic and anthelmintic properties.21,22Mangifera indica leaves, especially, show excellent anti-oxidant activity owing to the presence of the phenolic compounds such as mangiferin (a special xanthone termed a “super-anti-oxidant”) and quercetin, which have been widely studied for their pharmacological properties.23,24 Hence, these leaf extracts may provide green alternatives to replace the toxic reducing agents for reduction of GO.
On the other hand, modern devices such as touch screens, displays, organic light-emitting diodes (OLEDs) and photovoltaic cells have an essential component, transparent conducting film (TCF).25 In particular, graphene-based TCFs may provide a potential alternative to the conventional TCFs based on scarce and costly metals such as indium.26 Owing to its low cost, easy processing, and excellent dispersibility in organic solvents such as N,N-dimethylformamide (DMF), N,N-dimethylacetamide (DMA), and N-methyl-2-pyrrolidone (NMP) etc., solution-processed graphene synthesized by chemical reduction of GO has been investigated for TCF applications.27 For example, Li et al. have used a layer-by-layer deposition technique with exfoliated graphite in DMF solvent to obtain sheet resistance (Rs) and transmittance (T) of ∼150 kΩ □−1 and ∼92%, respectively, for the fabricated TCFs.28 Mattevi et al. have employed a transfer printing technique to fabricate TCFs with Rs of ∼3.5 kΩ □−1 and T of ∼85%.29 Similarly, Wu et al. have fabricated TCFs with Rs of ∼5 kΩ □−1 and T of ∼80% by a spin coating technique,30 while Zhao et al. have used a dip coating technique to fabricate TCFs with Rs and T of ∼8 kΩ □−1 and ∼70%, respectively.31 In addition, Sundramoorthy et al. have deposited TCFs by a vacuum filtration technique to attain Rs ∼ 890 ± 47 kΩ □−1 and T ∼ 90%.32 Recently, Lux et al. have presented a reliable process to deposit TCFs by a vacuum arc system on silicon oxide, quartz, and sapphire using a solid carbon source and have obtained Rs of 5.7 kΩ □−1 and T of 88%.33 Apart from these processes, spray coating is an effective technique for the deposition of TCFs of small grain size graphene platelets synthesized by a solution-processable method for better optoelectrical signatures.
Herein, Gns have been synthesized successfully with the help of leaf extracts at the low temperature of ∼50 °C. Leaf extracts from three different types of leaves, namely Ficus religiosa, Mangifera indica, and Polyalthia longifolia, have been used for reduction of GO. These are used as green reducing agents owing to their great anti-oxidant properties by virtue of the high content of phenolic compounds. Experimental results show that the maximum reduction occurred for the Mangifera indica leaf extract. This approach has several advantages such as low processing temperature, accessibility of the plant leaves and the non-toxic/environmentally friendly nature of the reducing agents (Table S1; ESI†). The procedure has been found more suitable for the production of good quality Gns at large scale. The as-synthesized Gns have been characterized by Raman spectroscopy, Fourier transform infrared (FTIR) spectroscopy, thermal gravimetric analysis (TGA), X-ray diffraction (XRD), UV-visible spectroscopy, field emission scanning electron microscopy (FESEM), and X-ray photoelectron spectroscopy (XPS). The synthesized Gns have been used further to fabricate TCFs by a simple spray coating technique (schematically shown in Fig. 1). Finally, optoelectrical properties have been measured after thermal graphitization of the TCFs. The best performing TCF shows a sheet resistance of ∼2.08 kΩ □−1 and a transmittance of 57.31%.
 | ||
| Fig. 1 Schematic of various steps in the fabrication process of graphene TCF. | ||
2. Experimental
2.1 Materials
Three different leaves, i.e., Ficus religiosa, Mangifera indica, and Polyalthia longifolia, were collected from I.I.T. Kanpur campus for leaf extract preparation. Graphite powder obtained from Loba Chemie Pvt Ltd, India, and sulfuric acid (H2SO4, 98%), hydrogen peroxide (H2O2, 30%), sodium nitrate (NaNO3, analytical grade), potassium permanganate (KMnO4, analytical grade) and N,N-dimethylformamide (DMF) procured from Qualigens Fine Chemicals, India, were used without further purification.2.2 Synthesis of GO
GO was synthesized by a modified Hummers' method as reported elsewhere.34 In brief, H2SO4 (140 ml), NaNO3 (2.5 g) and graphite powder (5 g) were mixed together, with cooling in an ice bath. Further, KMnO4 (15 g) was added to the mixture, maintaining the temperature below 20 °C till the mixture changed to green in color. Then, the mixture was shifted to a water bath and again stirred at 35 °C for 0.5 h. After adding 230 ml of deionized (DI) water dropwise, the mixture was stirred for 0.5 h, then the temperature was raised to 98 °C. Further, DI water (500 ml) and H2O2 (50 ml) were added until bubble formation stopped, and the mixture was stirred for 0.5 h. Finally, the resultant mixture was allowed to cool at room temperature. The solution was filtered and washed several times with DI water and dried at 60 °C overnight in ambient conditions.2.3 Preparation of leaf extract
Ficus religiosa, Mangifera indica, and Polyalthia longifolia leaves were collected and dried in the shade for 2 weeks before use. Then, 5 g chopped dry leaves of each were mixed in 200 ml of DI water, held at 50 °C for 1 h and allowed to cool to room temperature. Resultant mixtures were filtered to obtain the Ficus religiosa leaf extract (FLx), Mangifera indica leaf extract (MLx), and Polyalthia longifolia leaf extract (PLx), which were further used for the GO reduction.2.4 Reduction of GO
GO (0.1 mg ml−1) was mixed into 200 ml each of FLx, MLx and PLx and ultrasonicated at 100 W for 2 h to get a homogenous dispersion of GO. Once GO was fully dispersed in the leaf extracts, the resultant solutions were directly placed into an oil bath with continuous stirring at 50 °C for 24 h. Then, the products were washed several times with DI water and dried at 60 °C for 3 h in a vacuum oven. The Gns synthesized from FLx, MLx and PLx were named FGns, MGns and PGns, respectively. For comparison, another sample without any reducing agent was prepared under similar processing conditions (DI water/GO mixture (0.1 mg ml−1) stirred for 24 h at 50 °C followed by drying for 3 h at 60 °C).2.5 Fabrication of TCFs
A spray coating technique35 was used to fabricate TCFs of synthesized MGns. MGn dispersions (10 ml) in DMF (3 and 5 mg ml−1) were prepared by stirring for 12 h at 80 °C. Then, the MGns/DMF dispersions were centrifuged at 3000 rpm for 0.25 h and the top 5 ml dispersion volume was sprayed onto a 25 × 25 mm2 quartz substrate. The coated films of 3 and 5 mg ml−1 MGns in DMF were named GF-1 and GF-2, respectively. Before coating, the quartz substrates were ultrasonically cleaned in DI water and subsequently acetone and then dipped into piranha solution (3:1) to make the quartz surface more adhesive. An airbrush system with 0.2 mm nozzle diameter was used in single action mode; the distance between substrate and tip of the nozzle was kept at 12 cm with carrier gas (N2) pressure of 2 bar. Spray deposition on quartz substrates was performed by toggling the N2 valve (2 ml min−1) and the MGns/DMF dispersion was atomized into small droplets which were carried by N2 toward the substrate preheated to 200 °C. The coated films were dried in a vacuum oven for 3 h at 150 °C. Further, thermal graphitization was done at 500 °C for 3 h and 900 °C for 1 h in 10−3 torr vacuum.
2.6 Characterizations
A Raman microscope from Horiba Scientific (LabRam Micro-Raman spectrometer, Jobin-Yvon HR 800 UV) with He–Ne (633 nm) laser was used to characterize GO, FGns, MGns and PGns. FTIR spectroscopy of GO, FGns, MGns and PGns was carried out by a PerkinElmer Spectrum. XRD patterns of graphite powder, GO and MGns were recorded with X'Pert Powder PANalytical, advanced X-ray diffractometer with Cu Kα. UV-vis-NIR (near infrared) spectroscopy of GO and MGns was carried out by a PerkinElmer Lambda 1050. The surface morphologies of GO and MGns were examined by FESEM (JEOL JSM-7100F), while an atomic force microscope (AFM: Agilent 5500) was employed to study the surface roughness of as-synthesized MGns. Transmission electron microscopy (TEM: FEI Titan G2 60-300) was employed to study the structure of MGns further. TGA was carried out in a N2 atmosphere at a heating rate of 10 °C min−1 by a PerkinElmer Diamond TG/DT analyser. XPS measurements were carried out using a multifunctional XPS (ULVAC, PHI500 VersaProbe II with Al Kα (1486.6 eV) X-ray source). Electrical properties of fabricated TCFs were measured by a four-probe setup with PID-controlled oven (Scientific Equipment Roorkee, India) equipped with Keithley 6221, Keithley 2182A current and voltage sources, respectively. Optical properties were obtained in the visible range by PerkinElmer Lambda 1050. The film thickness (t) and surface roughness (Rz) were measured by a BRUKER (Contour GTK0X-14-150) optical profilometer.3. Results and discussion
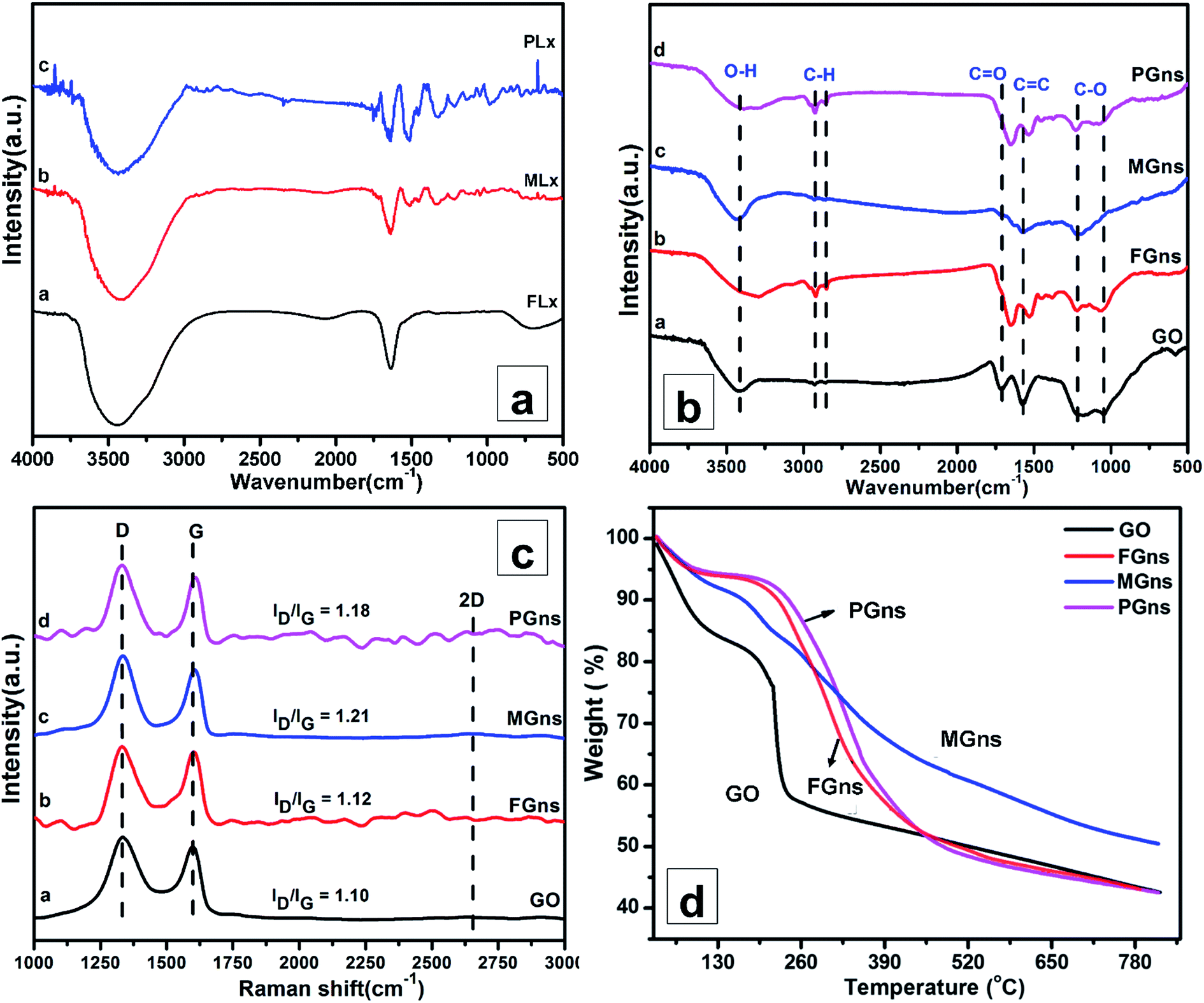
FTIR spectra of leaf extracts (FLx, MLx and PLx) are shown in Fig. 2a. The FTIR spectrum of FLx shows the 3436 cm−1 OH stretching of α-cellulose, 1635 cm−1 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching of lignin, and 687 cm−1 C–H stretching (β-glycosidic linkages between the monosaccharides). Similarly, the FTIR spectrum of MLx includes the mangiferin stretching vibration peak at 3399 cm−1 corresponding to the secondary O–H bond. The peak at 2903 cm−1 shows the presence of C–H anti-symmetric stretching, while the peaks at 1637, 1451 and 1324 cm−1 indicate the presence of C–O stretching, CH–CH bending and the C–O bond, respectively. Further, the FTIR spectrum of PLx shows the O–H stretching vibration peak at 3435 cm−1, amide I stretching vibration peak at 1638 cm−1, amide II stretching vibration peak at 1046 cm−1 and C–N stretching vibration of amine peak at 1046 cm−1.
C stretching of lignin, and 687 cm−1 C–H stretching (β-glycosidic linkages between the monosaccharides). Similarly, the FTIR spectrum of MLx includes the mangiferin stretching vibration peak at 3399 cm−1 corresponding to the secondary O–H bond. The peak at 2903 cm−1 shows the presence of C–H anti-symmetric stretching, while the peaks at 1637, 1451 and 1324 cm−1 indicate the presence of C–O stretching, CH–CH bending and the C–O bond, respectively. Further, the FTIR spectrum of PLx shows the O–H stretching vibration peak at 3435 cm−1, amide I stretching vibration peak at 1638 cm−1, amide II stretching vibration peak at 1046 cm−1 and C–N stretching vibration of amine peak at 1046 cm−1.
 | ||
| Fig. 2 FTIR spectra of (a) leaf extracts (FLx, MLx and PLx) and (b) GO and Gns (FGns, MGns and PGns). (c) Raman spectra and (d) TGA plots of GO and Gns (FGns, MGns and PGns). | ||
Essentially, the FTIR spectra of leaf extracts reveal the presence of various functional groups associated with the constituent phytochemicals. For example, MLx, the most active reducing agent among the studied leaf extracts, contains various phenolic compounds such as acetaldehyde (1.5%), furfural (0.6%), 2-furanomethanol (0.8%), phenol (3.1%), 2,3-dihydro-3,5-dihydroxy-6-methyl-4H-pyran-4-one (5.1%), o-catechol (1.3%), hydroquinone (1.7%), pyrogallol (26.9%), 2-hydroxyacetophenone (1.0%), diethyl phthalate (4.9%) and oleic acid (29.1%).36 Among these, pyrogallol, diethyl phthalate, hydroquinone, etc., possibly formed by the decomposition of mangiferin, are some of the known reducing agents present in the MLx.18,19 These reducing agents help in the removal of the oxygen-containing functional groups from GO during the reduction process in order to restore electronic conjugation in the honeycomb lattice.
Fig. 2b shows the FTIR spectra of GO, and of FGns, MGns and PGns produced by the leaf extracts. The FTIR spectrum of GO shows an intense broad peak at 3424 cm−1, which is attributed to an in-plane hydroxyl group, and a peak at 1717 cm−1, which is assigned to the CO stretching vibration of the carboxyl groups that are situated at the edges of GO sheets. The peaks at 1576, 1233 and 1039 cm−1 are associated with the CC, C–O (epoxy) and C–O (alkoxy) bonds, respectively.18,37 The FTIR spectrum of MGns shows reduced intensity of the peaks at 2926 and 2859 cm−1, corresponding to stretching vibrations of C–H bonds. Moreover, the peaks at 1717 and 1039 cm−1 have been suppressed after reduction, indicating the removal of most of the C–H and CO/C–O oxygen species from GO. Meanwhile, the FTIR spectra of FGns and PGns do not show significant suppression in the peaks corresponding to the oxygen-containing functional groups, which indicates partial removal of oxygen species from GO. Hence, the MLx produces the greater loss to C–H and C–O/CO groups, suggesting that MLx is the most effective green reducing agent for the preparation of Gns from GO.
Fig. 2c shows the Raman spectra of GO, FGns, MGns and PGns. The spectrum of GO exhibits two sharp intense peaks at 1332 and 1595 cm−1, which correspond to the D, G, and less intense 2D peak at 2629 cm−1, respectively. The G band at 1595 cm−1 corresponds to the in-plane vibration of sp2-bonded carbon atoms in the honeycomb lattice. The D band at 1332 cm−1 corresponds to disordered graphite; and arises due to the unsatisfied Raman fundamental selection rule of zone-boundary phonons,37 whereas the less intense peak at 2629 cm−1 corresponds to the 2D peak; and arises due to the second order zone boundary phonons. As compared with GO, a significant increment in the D to G band intensity ratio (ID/IG) is observed in MGns and PGns. Again, no change is observed in the Raman spectrum of the GO sample processed without reducing agent (Fig. S1, ESI†). This confirms the reduction of GO to be associated solely with the presence of the leaf extracts and not with the prolonged stirring in DI water at 50 °C. The increased ID/IG in MGns and PGns indicates the removal of oxygen functional groups from GO by the MLx and PLx, respectively. In particular, the MGn spectrum has a maximum value of ID/IG ∼ 1.21, which confirms maximum reduction of GO by MLx. Increased ID/IG ratio reveals the re-establishment of the conjugated graphene network (sp2 carbon) and signifies that more graphene domain has been formed during reduction, which results in a decrease in the average domain size.11,37 The Tuinstra–Koenig (TK) relation: La (nm) = (2.4 × 10−10) × λ4 × (ID/IG)−1 is used to calculate the crystalline domain size (La), and calculated values decrease from 34.71 nm for GO to 32.11 nm for MGns.
The thermal stability of as-prepared FGns, MGns and PGns has been assessed by TGA. TGA plots of GO, FGns, MGns and PGns are shown in Fig. 2d. The TGA plot of GO shows that ∼13.0 wt% mass loss occurs at below 100 °C, which is ascribed to the loss of water molecules adsorbed on and in the interlayer space of the GO sheets. Above 100 °C, degradation starts at 167 °C, mainly due to the loss of hydroxyl, epoxy functional groups and burning of aromatic carbons; ∼24.8 wt% of mass loss is observed up to 243 °C. A total weight loss of ∼56.7 wt% takes place below 800 °C. The TGA plot of FGns shows that the mass loss corresponding to adsorbed water (below 100 °C) is ∼5.9 wt%. Above 100 °C, degradation starts at 190 °C, mainly due to the loss of oxygen functionalities, and mass loss of ∼41.6 wt% has occurred up to 456 °C. On the other hand, PGns show higher thermal stability with a degradation onset point as high as 210 °C and mass loss of ∼41.0 wt% up to 456 °C, which indicates a higher extent of reduction of oxygen functional groups from GO by PLx. In addition, the TGA plot of MGns shows that ∼5.6 wt% of mass loss occurs at below 100 °C. Above 100 °C, gradual mass loss is observed mainly due to the degradation of hydroxyl and epoxy functional groups and burning of aromatic carbons. The total weight loss is observed to be ∼49.0 wt% up to 800 °C. The absence of a sharp degradation onset point in MGns indicates the smallest amounts of oxygen functionalities as compared with the FGns and PGns samples. Owing to the superior reduction activity of MLx, the MGns have the least amounts of oxygen functional groups. Hence, further characterizations have been carried out for the MGns sample only.
The XRD patterns of graphite powder, GO and MGns shown in Fig. 3a reveal their crystalline structures. The XRD pattern of graphite powder exhibits a sharp intense peak at 2θ of 26.55°, corresponding to the (002) reflection of hexagonal graphite (dspacing ∼ 3.34 Å).12,13 Similarly, the XRD pattern of GO shows that the graphite crystalline peak vanishes; and arises at 2θ of 12.19° (dspacing ∼ 7.22 Å),12,13 which indicates oxidation of graphite and presence of oxygen species in the GO. GO treated under similar conditions without leaf extract exhibits no change in the XRD pattern (Fig. S2, ESI†). After reduction by MLx at 50 °C, the sharp peak (001) at 2θ of 12.19° of GO disappears; and a broad peak at 2θ of 26.32 (dspacing ∼ 3.36 Å) appears. This signifies the exfoliation of the layered structure of GO and indicates the removal of oxygen functional groups from GO as well as formation of honeycomb lattice.38 Furthermore, the UV-visible spectra of GO and MGns are shown in Fig. 3b. The UV-vis spectrum of GO shows an absorption peak at 230 nm for GO, which is red shifted to 266 nm for MGns. The red shifting is attributed mainly to the change in electronic configuration of Gns during the process of reducing GO. Additionally, the absorption peak at 230 nm is ascribed to π–π* transition of the aromatic C–C ring. The red-shifted peak at 266 nm is ascribed to the n–π* transition of C–O bonds embedded in the MGns.38
 | ||
| Fig. 3 (a) XRD patterns of graphite powder, GO and MGns. (b) UV-vis spectra of GO and MGns. | ||
Surface morphological analysis was carried out by FESEM. Fig. 4a depicts the microstructure of GO, revealing randomly crumpled thin sheets of oxidized graphene closely associated with each other. Ultrasonication followed by mild heat-treatment in the presence of MLx reduces GO to Gns. The FESEM image in Fig. 4b shows the surface topography of the MGns, revealing the presence of multiple layers. The presence of seams on the surface is attributed to the overlapping/joining of different Gns. The TEM images of MGns shown in Fig. 4c and d reveal their crumpled structure consisting of multiple Gns entangled with each other to form a highly porous structure. The cylindrical structures in Fig. 4d are possibly formed by rolling up of individual Gns. The AFM image in Fig. 5a further reveals the surface topography of MGNs. Similar to the seam structures observed in the FESEM image Fig. 4b, the AFM image reveals the rough surface morphology of the Gns with typical height variations ranging between ±10 nm (inset: Fig. 5b).
 | ||
| Fig. 4 FESEM images of (a) GO and (b) MGns, and (c and d) TEM images of MGns. | ||
 | ||
| Fig. 5 (a) AFM image of MGns showing surface topography of Gns. (b) 3D view of (a). Inset of (b) shows the height profile near the middle of the AFM image (a). | ||
XPS survey spectra of the GO and MGns shown in Fig. 6a reveal their surface chemical state. The presence of the O 1s peak along with the C 1s peak in the XPS survey spectrum of GO (Fig. 6a) confirms harsh oxidation of graphite to GO. The O 1s peak intensity is decreased significantly after the reduction, indicating the formation of Gns. The high resolution C 1s spectrum of GO, deconvoluted by using Voigt distributions and Shirley type background (Fig. 6b), consists of two intense peaks corresponding to C–C and C–O bonds of carbon atoms in a 2D planner lattice at binding energies of 284.6 and 286.6 eV, respectively, indicating a considerably high degree of oxidation. Further, weak peaks at binding energies of 287.1 and 288.9 eV are attributed to the CO and C–CO carbon bonding configurations, indicating harsh oxidation and destruction of the sp2 atomic structure of graphite.39 The intensities of C–O, CO and C–CO peaks decrease drastically when GO is reduced by MLx at 50 °C to produce MGns (C/O ratio increases from 2.22 to 4.58), which strongly indicates the restoration of π delocalized conjugation in the honeycomb lattice (Fig. 6c).40 In addition, the high resolution O 1s spectrum of MGns shown in Fig. 6d clearly indicates that the spectrum consists of two peaks at binding energies of 532.6 and 533.7 eV, which can respectively be assigned to C–O and CO absorbed on the surface of the honeycomb network. XPS of MGns does not detect any impurity peak, confirming the formation of high quality Gns through reduction of GO by MLx. The structural characterizations through Raman, FTIR spectroscopy, SEM, TEM and XPS confirm that MLx is an excellent green reducing agent for reduction of GO to produce Gns at large scale.
 | ||
| Fig. 6 XPS analysis. (a) XPS survey of GO and MGns. (b) High resolution spectra C 1s of GO. (c) High resolution spectra C 1s of MGns. (d) High resolution spectra O 1s of MGns. | ||
Fig. 7 shows the optoelectrical signatures of MGns films deposited on quartz substrates by spray coating. The optoelectrical properties of the coated films were measured after thermal graphitization of GF-1 and GF-2 at 500 °C for 3 h. The photographic images of the fabricated films are shown in the inset of Fig. 7a. It was observed that the average transmittance (Tav) values in the visible light region (380 to 780 nm) are ∼45.48 and 32.96% for GF-1 and GF-2, respectively, while the transmittance (T) at 550 nm was found to be ∼44.34 and ∼32.40% for the respective samples (Fig. 7a). In addition, the Rs values of the coated films were found to be ∼82.44 and 92.24 kΩ □−1 for GF-1 and GF-2, respectively. Furthermore, the temperature dependent sheet resistance behavior of GF-1 and GF-2 was investigated in the temperature range from 25 to 180 °C (Fig. 7b) by using a hot air oven model with attached Keithley current–voltage source. The result showed that the Rs value decreased with temperature, possibly due to increased ionization states of impurities and/or increased charge carrier mobility. The decrease in Rs values of Gns film with increasing temperature is in agreement with the theoretical calculations by Fang et al.41 In addition, to investigate the effect of the thermal graphitization on optoelectrical signatures; GF-1 and GF-2 were annealed further at 900 °C for 1 h and it was observed that after thermal graphitization the Tav was increased to 57.6 and 35.3% for GF-1 and GF-2, respectively (Fig. 7c). The photographic images of annealed films are shown in the inset of Fig. 7c. Meanwhile, T at 550 nm wavelength was found to be ∼57.3 and 35.7% for GF-1 and GF-2, respectively, whereas the Rs of coated films decreased to 2.08 and 6.88 kΩ □−1 for GF-1 and GF-2, respectively. The temperature dependent sheet resistance behavior of GF-1 and GF-2 is shown in Fig. 7d.
 | ||
| Fig. 7 Optoelectrical signatures of films. (a) Transmittance of GF-1 and GF-2 films. Inset: photographs of films. (b) Sheet resistance vs. temperature of GF-1 and GF-2 films, annealed at 500 °C. (c) Transmittance of GF-1 and GF-2 films Inset: photograph of GF-1 and GF-2 films. (d) Sheet resistance vs. temperature of GF-1 and GF-2, annealed at 900 °C. | ||
To determine the influence of thermal graphitization on sp2 domains, structural studies of GF-1 and GF-2 were carried out by using Raman spectroscopy and XRD analysis. Fig. 8a shows the Raman spectra of GF-1 and GF-2 films, exhibiting prominent spectral features such as D bands at 1346 and 1348 cm−1 and G bands at 1610 and 1608 cm−1 for GF-1 and GF-2, respectively, when films were annealed at 500 °C for 3 h, while the respective ID/IG ratios were found to be ∼1.39 and 1.26. When the annealing temperature was increased to 900 °C, red shifting was observed in the D and G bands. For CF-1 and CF-2, the D bands were observed at 1350 and 1352 cm−1, respectively, while the corresponding G bands lie at 1614 and 1612 cm−1. The ID/IG ratios were found to be ∼1.27 and 1.23 for GF-1 and GF-2, respectively, significantly lower than those for the lower annealing temperatures. This may be attributed to the removal of oxygen species from the carbon skeleton as well as the clustering of the sp2 phase and forming of a more ordered structure upon annealing.42
 | ||
| Fig. 8 Structural study of GF-1 and GF-2 films at different annealing temperature. (a) Raman analysis. (b) XRD analysis. | ||
Furthermore, crystalline structures of the coated films were analyzed by XRD patterns, shown in Fig. 8b. For an annealing temperature of 500 °C, the dominant peak in the XRD pattern was observed at 2θ of 26.33° and 26.37° for GF-1 and GF-2, respectively. The respective interlayer distances (dspacing) were calculated to be ∼3.367 and ∼3.362 Å for GF-1 and GF-2. On the other hand, the most intense diffraction peak shifts towards higher angle with increase in annealing temperature (900 °C for 1 h) and is found at 2θ of 26.39° (dspacing ∼ 3.360 Å) and 26.41° (dspacing ∼ 3.357 Å) for GF-1 and GF-2, respectively. These dspacing values are close to the reported interlayer distance values for graphene (∼3.354 Å), which confirms the restoration of sp2 domains by thermal graphitization.43 Thus, it is evident from the structural studies of films that restoration of the sp2 carbon network occurs with increase in annealing temperature and more linkages are formed between sp2 carbon networks. This indicates that the film structure shifts towards the graphene structure and the possibility of degradation of graphene film can be easily discarded. Hence, the transmittance of the film increases, Rs of the film decreases and electrical conductively increases. Electrical conductively of films44 was found to be ∼18.8 and ∼3.6 S cm−1 for GF-1 and GF-2, respectively. To evaluate the performance of the fabricated TCFs, a figure of merit (FOM), defined as 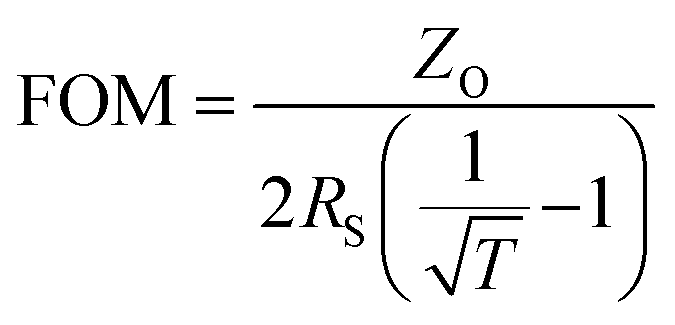 , where ZO is the free space impedance (377 Ω), has been calculated based on Rs and T, and a high value of FOM signifies the better performance of TCFs, which requires high transparency and low sheet resistance.45 The performance evaluations of GF-1 and GF-2 are tabulated in Table 1 and it is seen that GF-1 shows better performance as TCFs than GF-2. The comparison of optoelectrical properties of graphene-based TCFs deposited by different processes are tabulated in Table 2.
, where ZO is the free space impedance (377 Ω), has been calculated based on Rs and T, and a high value of FOM signifies the better performance of TCFs, which requires high transparency and low sheet resistance.45 The performance evaluations of GF-1 and GF-2 are tabulated in Table 1 and it is seen that GF-1 shows better performance as TCFs than GF-2. The comparison of optoelectrical properties of graphene-based TCFs deposited by different processes are tabulated in Table 2.
| Film name | t a, nm | Annealed 500 °C for 3 h | Annealed 900 °C for 1 h | ||||||
|---|---|---|---|---|---|---|---|---|---|
| R s a, kΩ □−1 | T a, % | σ a, S cm−1 | FOMa × 10−3 | R s a, kΩ □−1 | T a, % | σ a, S cm−1 | FOMa | ||
| a t, thickness of film; Rs, sheet resistance; T, transmittance at 550 nm; σ, conductivity; FOM, figure of merit. | |||||||||
| GF-1 | 255 | 82.4 | 44.3 | 0.5 | 4.5 | 2.1 | 57.3 | 18.8 | 0.28 |
| GF-2 | 401 | 92.2 | 32.4 | 0.3 | 2.7 | 6.9 | 35.7 | 3.6 | 0.04 |
| Method | Precursor material | Reducing agent | Reducing agent issue | T (%) | R s (kΩ □−1) | Ref. |
|---|---|---|---|---|---|---|
| L–B deposition | GO | Thermal reduction | — | 92.0 | 150.0 | 28 |
| Transfer printing | GO | N2H4 | Toxic | 85.0 | 3.5 | 29 |
| Spin coating | GO | N2H4 | Toxic | 80.0 | 5.0 | 30 |
| Dip coating | GO | Thermal reduction | — | 70.0 | 8.0 | 31 |
| Vacuum filtration | GO | N2H4 | Toxic | 90.0 | 890.0 | 32 |
| Transfer printing | GO | N2H4 | Toxic | 80.0 | 2.2 | 46 |
| Dip coating | GO | N2H4 | Toxic | 80.0 | 114.0 | 47 |
| Spin coating | GO | N2H4 | Toxic | 89.0 | 1.1 | 48 |
| Drop cast | GO | HI | Toxic | 70.0 | 0.2 | 49 |
| Spin coating | GO | N2H4 | Toxic | 78.0 | 3.1 | 50 |
| Rod coating | GO | Sn | Not toxic | 75.0 | 6.7 | 51 |
| Spray coating | GO | Leaf extract | Natural | 57.31 | 2.1 | Present work |
Finally, surface roughness analysis of TCFs was carried out by optical profiler. It was observed that the films were coated uniformly over the quartz substrate; and the surface average roughness (Rz) was found to be ∼10.2 and ∼21.0 μm for GF-1 and GF-2, respectively (Fig. 9a and b). In addition, the thickness (t) of coated films was measured; and found to be ∼255 and 401 nm for GF-1 and GF-2, respectively. It was also observed that Rz and t values increased gradually with the concentration of MGns. The comparison of transparency and conductivity of graphene-based TCFs deposited by spray coating using different reducing agents is tabulated in Table 3. The results show that the best performing graphene-based TCFs were deposited by a simple spray coating approach from solution-processable Gns synthesized via green reduction of GO in the presence of MLx; and achieved a sheet resistance ∼2.1 kΩ □−1 with a transmittance of 57.3%, and had a FOM value ∼0.28.
 | ||
| Fig. 9 Surface roughness images, 2D and 3D, of (a) GF-1 and (b) GF-2 films annealed at 900 °C. | ||
| Method | Precursor material | Reducing agent | Reducing agent issue | T (%) | R s (kΩ □−1) | Ref. |
|---|---|---|---|---|---|---|
| Spray coating | GO | N2H4 | Toxic | 84.0 | 2.2 | 35 |
| Spray coating | GO | HI | Toxic | 74.0 | 15.3 | 52 |
| Spray coating | GO | N2H4 | Toxic | 68.7 | 58.0 | 53 |
| Spray coating | GO | N2H4 | Toxic | 80.0 | — | 54 |
| Spray coating | GO | N2H4 | Toxic | 80.0 | 0.3 | 55 |
| Spray coating | GO | N2H4 | Toxic | 72.7 | 19.0 | 56 |
| Spray coating | GO | N2H4 | Toxic | 96.0 | 20.0 | 57 |
| Spray coating | GO | Leaf extract | Natural | 57.3 | 2.1 | Present work |
4. Conclusions
In summary, we have demonstrated a simple green reduction approach for GO to produce Gns using FLx, MLx and PLx. The reductive abilities of FLx, MLx and PLx were analyzed at 50 °C. Raman analysis affirmed that the degree of reduction of GO was a maximum for MLx, with an ID/IG ratio of ∼1.21. FTIR analysis validated the Raman signature of maximum removal of oxygen species from GO by MLx. XPS analysis confirmed the removal of oxygen functional groups from GO, with a maximum C/O ratio of ∼4.58. This signifies that the MLx offers a potential alternative to avoid the use of hazardous reducing agents and thus minimize environmental impact by green synthesis of Gns at large scale. Furthermore, TCFs were deposited by simple spray coating of synthesized MGns, and their optoelectrical properties were measured. The Rs and T values of the best-performing GF-1 TCF were found to be ∼92.2 kΩ □−1 and ∼44.3%, respectively, for the annealing temperature of 500 °C (3 h), while for the GF-1 annealed at 900 °C for 1 h, Rs decreased significantly to ∼2.1 kΩ □−1 with an increase of T to ∼57.3%.Acknowledgements
The authors acknowledge the financial support provided by the Indian Institute of Technology Kanpur, India for carrying out this research work.References
- K. S. Novoselov, V. I. Falko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22(35), 3906–3924 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332(6037), 1537–1541 CrossRef CAS PubMed.
- Y. Zhao, X. Li, Y. Du, G. Chen, Y. Qu, J. Jiang and Y. Zhu, Nanoscale, 2014, 6, 11112–11120 RSC.
- K. K. Kar, S. Rana and J. K. Pandey, Handbook of Polymer Nanocomposites. Processing, Performance and Application, Volume B: Carbon Nanotube Based Polymer Nanocomposites, Springer, Heidelberg, New York, Dordrecht, London, 2015 Search PubMed.
- Y. Chen, C. Tan, H. Zhang and L. Wang, Chem. Soc. Rev., 2015, 44, 2681–2701 RSC.
- K. Turcheniuk, R. Boukherroub and S. Szunerits, J. Mater. Chem. B, 2015, 3, 4301–4324 RSC.
- K. K. Kar and A. Hodzic, Developments in Nanocomposites, Research Publishing Services, Singapore, 2014 Search PubMed.
- S. M. Kim, J. H. Kim, K. S. Kim, Y. Hwangbo, J. H. Yoon, E. K. Lee, J. Ryu, H. J. Lee, S. Cho and S. M. Lee, Nanoscale, 2014, 6, 4728–4734 RSC.
- P. Chamoli, M. K. Das and K. K. Kar, Graphene, 2015, 3, 56–60 CrossRef.
- S. Pei and H. M. Cheng, Carbon, 2012, 50(9), 3210–3228 CrossRef CAS.
- Z. Xiong, T. Gu and X. Wang, Langmuir, 2014, 30(2), 522–532 CrossRef CAS PubMed.
- Z. Xiong, C. Liao and X. Wang, J. Mater. Chem. C, 2015, 3, 6224–6231 RSC.
- Z. Xiong, C. Liao, W. Han and X. Wang, Adv. Mater., 2015, 27(30), 4469–4475 CrossRef CAS PubMed.
- Z. Xiong, X. Yun, B. Tang and X. Wan, Carbon, 2016, 107, 548–556 CrossRef CAS.
- Z. Xiong, C. Liao and X. Wang, J. Mater. Chem. A, 2014, 2, 19141–19144 CAS.
- M. Agharkar, S. Kochrekar, S. Hidouri and M. A. Azeez, Mater. Res. Bull., 2014, 59, 323–328 CrossRef CAS.
- S. Thakur and N. Karak, Carbon, 2012, 50(14), 5331–5339 CrossRef CAS.
- S. Barua, S. Thakur, L. Aidew, A. K. Buragohain, P. Chattopadhyay and N. Karak, RSC Adv., 2014, 4, 9777–9783 RSC.
- M. Nasrollahzadeh and S. Mohammad Sajadi, RSC Adv., 2015, 5, 46240–46246 RSC.
- C. P. Khare, Indian medicinal plants, Springer-Verlag, Berlin, Heidelberg, Germany, 2007 Search PubMed.
- N. Bhatla, T. Mukharjee and G. Singh, Indian J. Hist. Sci., 1984, 19(1), 37–42 Search PubMed.
- M. Masibo and Q. He, Compr. Rev. Food Sci. Food Saf., 2008, 7(4), 309–319 CrossRef.
- C. G. Mohan, D. Mundkinajeddu, G. L. Viswanath, G. Savinay, V. Hanumantharaju, C. E. Rajendra and P. D. Halemani, Asian Pac. J. Trop. Med., 2013, 6(4), 311–314 CrossRef CAS PubMed.
- K. Ellmer, Nat. Photonics, 2012, 6, 809–817 CrossRef CAS.
- J. K. Wasseia and R. B. Kaner, Mater. Today, 2010, 13(3), 52–59 CrossRef.
- J. I. Paredes, S. Villar-Rodil, A. Martınez-Alonso and J. M. D. Tascon, Langmuir, 2008, 24, 10560–10564 CrossRef CAS PubMed.
- X. Li, G. Zhang, X. Bai, X. Sun, X. Wang, E. Wang and H. Dai, Nat. Nanotechnol., 2008, 3, 538–542 CrossRef CAS PubMed.
- C. Mattevi, G. Eda, S. Agnoli, S. Miller, K. A. Mkhoyan, O. Celik, D. Mastrogiovanni, G. Granozzi, E. Garfunkel and Manish Chhowalla, Adv. Funct. Mater., 2009, 19, 2577–2583 CrossRef CAS.
- J. B. Wu, H. A. Becerril, Z. N. Bao, Z. F. Liu, Y. S. Chen and P. Peumans, Appl. Phys. Lett., 2008, 92, 263302 CrossRef.
- L. Zhao, L. Zhao, Y. X. Xu, T. F. Qiu, L. J. Zhi and G. Q. Shi, Electrochim. Acta, 2009, 55, 491–497 CrossRef CAS.
- A. K. Sundramoorthy, Y. Wang, J. Wang, J. Che, Y. X. Thong, A. C. W. Lu and M. B. Chan-Park, Sci. Rep., 2015, 5, 10716 CrossRef CAS PubMed.
- H. Lux, P. Siemroth, A. Sgarlata, P. Prosposito, M. A. Schubert, M. Casalboni and S. Schrader, J. Appl. Phys., 2015, 117, 195304 CrossRef.
- P. Chamoli, M. K. Das and K. K. Kar, Current Nanomaterials, 2016, 1(2), 110–116 CrossRef.
- V. H. Pham, T. V. Cuong, S. H. Hur, E. W. Shin, J. S. Kim, J. S. Chung and E. J. Kim, Carbon, 2010, 48, 1945–1951 CrossRef CAS.
- D. Martínez-Bernett, A. Silva-Granados, S. N. Correa-Torres and A. Herrera, J. Phys.: Conf. Ser., 2016, 687, 012033 CrossRef.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS PubMed.
- Y. Piao and B. Chen, RSC Adv., 2016, 6, 6171–6181 RSC.
- S. Some, Y. Kim, Y. Yoon, H. J. Yoo, S. Lee, Y. Park and H. Lee, Sci. Rep., 2013, 3, 1929 Search PubMed.
- D. Yang, A. Velamakanni, G. Bozoklu, S. Park, M. Stoller, R. D. Piner, S. Stankovich, I. Jung, D. A. Field, C. A. Ventrice Jr and R. S. Ruoff, Carbon, 2009, 47, 145–152 CrossRef CAS.
- X.-Y. Fang, X.-X. Yu, H.-M. Zheng, H.-B. Jin, L. Wang and M.-S. Cao, Phys. Lett. A, 2015, 379, 2245–2251 CrossRef CAS.
- S. J. Wang, Y. Geng, Q. Zheng and J. K. Kim, Carbon, 2010, 48(6), 1815–1823 CrossRef CAS.
- H. A. Becerril, J. Mao, Z. Liu, R. M. Stoltenberg, Z. Bao and Y. Chen, ACS Nano, 2008, 2(3), 463–470 Search PubMed.
- S. H. Tamboli, B. S. Kim, G. Choi, H. Lee, D. Lee, U. M. Patil, J. Lim, S. B. Kulkarni, S. C. Juna and H. H. Cho, J. Mater. Chem. A, 2014, 2, 5077–5086 Search PubMed.
- Q. Zheng, W. H. Ip, X. Lin, N. Yousefi, K. K. Yeung, Z. Li and J. K. Kim, ACS Nano, 2011, 5(7), 6039–6051 Search PubMed.
- S. Y. Jeong, S. H. Kim, J. T. Han, H. J. Jeong, S. Yang and G. W. Lee, ACS Nano, 2011, 5(2), 870–878 Search PubMed.
- S. Mihara, T. Tsubota, N. Murakami and T. Ohno, J. Nanosci. Nanotechnol., 2012, 12(9), 6930–6934 Search PubMed.
- R. Karthick, M. Brindha, M. Selvaraj and S. Ramu, J. Colloid Interface Sci., 2013, 406, 69–74 CrossRef CAS PubMed.
- A. Nekahi, P. H. Marashi and D. Haghshenas, Appl. Surf. Sci., 2014, 295, 59–65 CrossRef CAS.
- X. D. Betriu, S. A. García, C. Botas, P. Álvarez, J. S. Marcos, C. Prieto, R. Menéndezb and A. de Andrés, J. Mater. Chem. C, 2013, 1, 6905–6912 RSC.
- J. Ning, J. Wang, X. Li, T. Qiu, B. Luo, L. Hao, M. Liang, B. Wang and L. Zhi, J. Mater. Chem. A, 2014, 2, 10969–10973 CAS.
- X. Li, D. Zhang, C. Yang and Y. Shang, J. Nanosci. Nanotechnol., 2015, 15(12), 9500–9508 CrossRef CAS PubMed.
- H. Shi, C. Wang, Z. Sun, Y. Zhou, K. Jin and G. Yang, Sci. China: Phys., Mech. Astron., 2015, 58(1), 1–5 CrossRef CAS.
- K. Min, T. H. Han, J. Kim, J. Jung, C. Jung, S. M. Hong and C. M. Koo, J. Colloid Interface Sci., 2012, 383(1), 36–42 CrossRef CAS PubMed.
- Y. J. Lin, A. Zhamu, B. Z. Jang, S. Lee and J. C. Lin, US Pat., 20140272199 A1, 2014.
- D. Y. Kim, S. S. Ray, J. J. Park, J. G. Lee, Y. H. Cha, S. H. Bae, J. H. Ahn, Y. C. Jung, S. M. Kim, A. L. Yarin and S. S. Yoon, Adv. Funct. Mater., 2014, 24(31), 4986–4995 CrossRef CAS.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra19111h |
| This journal is © The Royal Society of Chemistry 2016 |