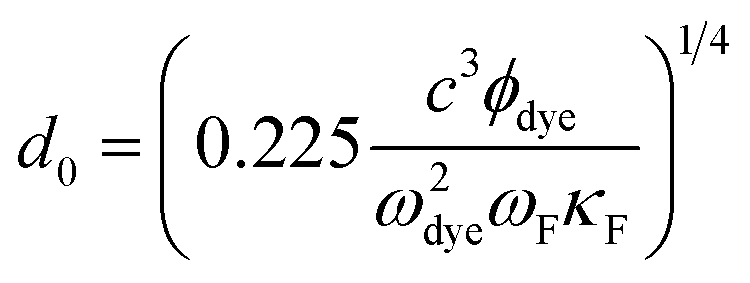DOI:
10.1039/C6RA18954G
(Paper)
RSC Adv., 2016,
6, 98753-98760
Direct observation of electronic transition–plasmon coupling for enhanced electron injection in dye-sensitized solar cells
Received
26th July 2016
, Accepted 8th October 2016
First published on 10th October 2016
Abstract
We illustrate experimental evidence of the effect of surface plasmon resonance (SPR) of a noble metal on the ultrafast-electron injection efficiencies of a sensitizing dye in proximity of a wide band gap semiconductor. We have compared the effect of Au with Al nanoparticles as the former have a strong SPR band (peak 560 nm) at the emission (∼600 nm) of the model dye protoporphyrin IX (PP) in the proximity of mesoporous TiO2 nanoparticles in a model dye sensitized solar cell (DSSC). We have used detailed electron microscopic procedures for the characterization of Au/Al nanoparticle-embedded TiO2, the host of PP. Picosecond resolved emission spectroscopy on the model dye reveals an ultrafast component consistent with photoinduced electron transfer (PET) from the dye to the TiO2 matrix in the presence of Au nanoparticles. In order to investigate the dipolar separation of PP from the Au nanoparticle surface, we have employed a Förster Resonance Energy Transfer (FRET) strategy in the PP–Au nanoparticle system in the absence of TiO2. Although the time scale of FRET and PET were found to be similar, the plasmon induced enhanced electron transfer in the case of Au nanoparticles is found to be clear from various device parameters of the plasmonic solar cell (DSSC) designed from the materials. We have also fabricated a DSSC with the developed materials consisting of Al–Au nanoparticles with N719 dye as sensitizer. The fabricated DSSC exhibits a much higher power conversion efficiency of (7.1 ± 0.1)% compared to that with TiO2 alone (5.63 ± 0.13)%. The outstanding performance of DSSC based on plasmonic nanoparticles was attributed to the plasmonic coupling and scattering effect for enhanced electron injection efficiencies.
1. Introduction
The mechanistic pathways for the enhancement of the efficiency of a dye sensitized solar cell (DSSC) using plasmonic nanoparticles have attracted lots of attention in contemporary literature.1–8 The specific role of plasmon in the nanoparticles over the nonspecific scattering of incident radiation leading to the ‘light trapping’ has been addressed in recent studies.9–13 In a theoretical investigation on a model DSSC, the magnitude of dipole–dipole coupling between the plasmon resonance of metallic nanoparticles and the electronic transition in the sensitizing dye has been carefully varied and an optimum coupling strength for which the electron population injected into the host semiconductor is maximized.14 The delocalization of plasmonic state in the proximity of wide band gap semiconductor in a DSSC, leading to conversion of photon to charge carriers has been predicted in another computational study.9 The role of spacing between the semiconductor surface and plasmonic metal structure on the rate of electron injection from metal to semiconductor has also been demonstrated experimentally.15,16 However, the direct experimental evidence of dipolar coupling between electronic transition of a dye in a DSSC and plasmonic resonance of a metal nanoparticle in active electrode on the overall efficiency of the DSSC is sparse in the existing literature, and is the motive of the present work.
Here we have studied protoporphyrin IX (PP) sensitized TiO2 as photoanode in DSSC as model solar cell. The semiconductor (TiO2) is decorated with Au nanoparticles having absorption maxima at 530 nm due to surface plasmon resonance (SPR) band, which has spectral overlap with the fluorescence of the model dye in the DSSC. We have utilized steady state and picosecond resolved photoluminescence of the dye PP in order to confirm dipolar coupling with the Au nanoparticles. The manifestation of the dipolar coupling in the photocurrent and photovoltage of the DSSC have also been investigated. In order to decouple the interference of non-specific light scattering by the Au nanoparticles on the enhancement of the DSSC efficiency, we have used synthesized aluminum nanoparticles (SPR at 370 nm) as scattering layer in the DSSC and compared enhancement of efficiency. A clear difference in the electron injection due to the SPR in the Al nanoparticles decorated DSSC is evident in our studies. We have fabricated DSSCs with plasmonic nanoparticles (Al and Au) which exhibit much higher power conversation efficiencies in compared to their counterparts. In order to validate the proposed mechanism of dipolar coupling for conventional dyes, we have used non-emissive N719 as sensitizing dye to fabricate DSSCs and found that it is following the similar trend as that of the PP with higher solar cell efficiencies.
2. Experimental section
2.1. Reagents
TiO2, protoporphyrin IX (PP), di-tetrabutylammonium cis-bis(isothiocyanato)bis(2,2′-bipyridyl-4,4′-dicarboxylato)ruthenium(II) (N719), gold(III)chloride trihydrate, platinum chloride (H2PtCl6), lithium iodide (LiI), iodine (I2) and 4-tert-butylpyridine (TBP) were purchased from Sigma-Aldrich. Ultrapure water (Millipore System, 18.2 MΩ cm) and ethanol (≥99% for HPLC, purchased from Sigma-Aldrich) were used as solvents. All other chemicals used in the study were of analytical grade and were used without further purification. Fluorine-doped tin oxide (FTO) conducting glass substrates, acquired from Sigma-Aldrich were cleaned by successive sonication with soap water, acetone, ethanol, and deionized (DI) water for 20 min, each with adequate drying prior to their use.
2.2. Synthesis of gold and aluminum modified TiO2
The Au–TiO2 nanohybrid was obtained by chemical reduction of Au(III) in the TiO2 suspension following simple one-step chemical reduction method.17,18 An aqueous solution of TiO2 suspension is sonicated for 20 min, then 5 μL of 100 mM HAuCl4 added under continuous starring for 5 min. After that 200 μL of 10 mM NaBH4 was added, the reaction was allowed to continue for another 30 min. The Au–TiO2 nanohybrid were centrifuged and dried at 80 °C.
The aluminum nanoparticles were synthesized following the previous report with some modification.19 At first 0.01 mmol of aluminum acetylacetonate [Al(acac)3] along with TiO2 were added to the toluene which was already placed in a two neck round-bottom flask (RBF) equipped with a magnetic stirring bar. Then, 0.03 mmol of lithium aluminum hydride (LiAlH4) was added into the reaction flask. The reaction mixture was then refluxed continuously with stirring approximately for 72 h under nitrogen atmosphere. After completion, the reaction mixture was cooled down to room temperature and a gray-colored precipitate was settled down. The crude product obtained was washed with ice cold methanol for several times in order to wash out organic part and the unreacted starting materials. Finally the product was dried at low pressure under a nitrogen sink. In order to synthesize the nanoparticles (Au/Al) without TiO2 matrix, we have followed above procedures excluding the addition of TiO2 nanoparticles in the procedures. We have conjugated PP with the synthesized nanoparticles by overnight stirring and repeated washing using dimethyl sulfoxide.
2.3. Fabrication of DSSCs
For the fabrication of DSSCs, at first TiO2 paste was coated on a FTO glass substrate. The ratio of the metal nanoparticles (Au and Al) to TiO2 could be readily adjusted by changing the concentration of the nanoparticles. These photoanode were annealed at 450 °C for 1 h. After that photoanode were immersed in a 0.5 mM PP and N719 solutions for 24 h at room temperature. For preparation of counter electrode, the platinum (Pt) was deposited on the FTO substrates by thermal decomposition of 10 mM platinum chloride (in isopropanol) at 385 °C for 30 min. The two electrodes were placed on top of each other with a single layer of 60 μm thick Surlyn (Solaronix) as a spacer between the two electrodes. A liquid electrolyte composed of 0.5 M lithium iodide (LiI), 0.05 M iodine (I2) and 0.5 M 4-tert-butylpyridine (TBP) in acetonitrile was used as the hole conductor and filled in the inter electrode space by using capillary force, through two small holes (diameter = 1 mm) predrilled on the counter electrode. Finally, the two holes were sealed by using another piece of Surlyn to prevent the leakage of electrolyte from the cell. In all our experiments, the active area of the DSSCs was fixed at 0.64 cm2.
2.4. Characterization methods
Field Emission Scanning Electron Microscopy (FESEM, QUANTA FEG 250) was used to investigate the surface morphology of the samples by applying a diluted drop of nanoparticles on a silicon wafer. Transmission Electron Microscopy (TEM) grids were prepared by applying a diluted drop of aluminum and (Au)TiO2 samples to carbon-coated copper grids. FEI (Technai S-Twin, operating at 200 kV) TEM instrument with a high magnification of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× was used for particle size determination from micrographs recorded. X-Ray Diffraction (XRD) patterns of the samples were obtained by a PANalytical XPERTPRO diffractometer equipped with Cu Kα radiation (at 40 mA and 40 kV) at a scanning rate of 0.02° S−1 in the 2θ range from 20° to 90°. The current density–voltage characteristics of the cells were recorded by Keithley under an irradiance of 100 mW cm−2 (AM 1.5 simulated illuminations, Photo Emission Tech). The wavelength-dependent photocurrent is measured using a homemade setup with a Bentham monochromator and dual light (tungsten and xenon) sources. Photovoltage decay measurements were carried out after illuminating the cells under 1 sun. The photovoltage decays after switching off the irradiation were monitored by an oscilloscope (Owon) through computer interface. The decays were fitted with exponential decay functions using origin software. For steady state and time resolved optical studies, we have followed the methodology as described in our earlier work.20,21 The Förster distance (R0) is given by,
000× was used for particle size determination from micrographs recorded. X-Ray Diffraction (XRD) patterns of the samples were obtained by a PANalytical XPERTPRO diffractometer equipped with Cu Kα radiation (at 40 mA and 40 kV) at a scanning rate of 0.02° S−1 in the 2θ range from 20° to 90°. The current density–voltage characteristics of the cells were recorded by Keithley under an irradiance of 100 mW cm−2 (AM 1.5 simulated illuminations, Photo Emission Tech). The wavelength-dependent photocurrent is measured using a homemade setup with a Bentham monochromator and dual light (tungsten and xenon) sources. Photovoltage decay measurements were carried out after illuminating the cells under 1 sun. The photovoltage decays after switching off the irradiation were monitored by an oscilloscope (Owon) through computer interface. The decays were fitted with exponential decay functions using origin software. For steady state and time resolved optical studies, we have followed the methodology as described in our earlier work.20,21 The Förster distance (R0) is given by,| | |
R0 = 0.211 × [κ2η−4QDJ]1/6
| (1) |
Once the value of R0 is known, the distance between the donor and acceptor (r) was calculated using the following equation,
where,
E is the efficiency of energy transfer and following the procedure published earlier.
22
The nanosurface energy transfer (NSET) model become useful when the rate of surface energy transfer is expected to follow 1/d4 distance dependence, where d is the distance of the energy donor from the nano-surface. The d0 value was calculated using NSET equation as follows:
| |
 | (3) |
where
ϕdye is the quantum yield of dye,
c is the speed of light,
ωdye is the angular frequency of dye,
ωF is the angular frequency of bulk gold,
κF is the rate of energy transfer and details of the procedure are discussed in our previous studies.
23
3. Results and discussion
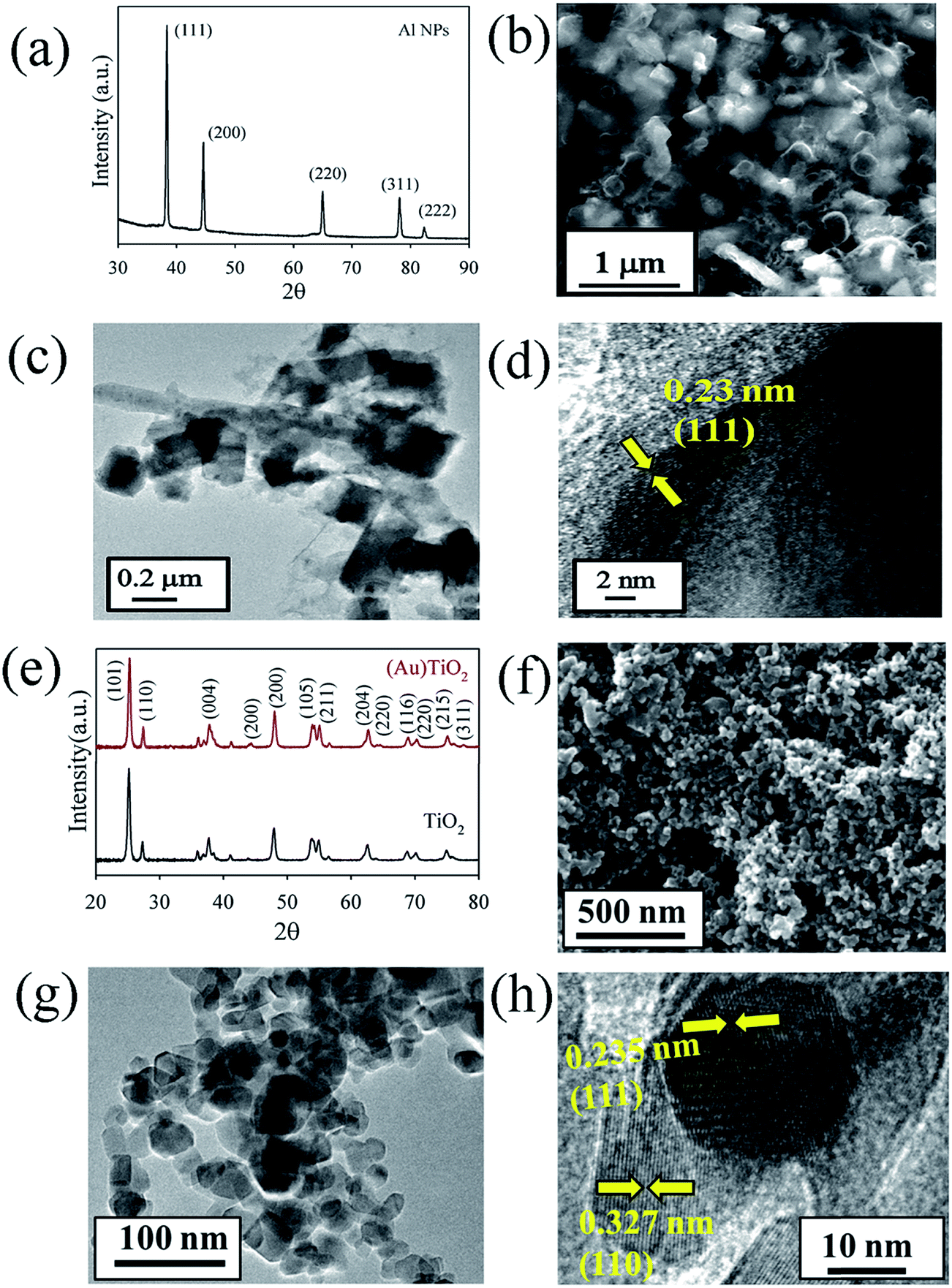
The powder X-ray diffraction pattern of as synthesized Al nanoparticles is shown in Fig. 1a which is compared with the JCPDS data (#04-0787) and found to be in good agreement with the face-centered cubic (fcc) form of aluminum. Fig. 1b shows the morphology of the synthesized Al nanoparticles as revealed from FESEM. The size of the Al nanoparticles is observed by HRTEM as shown in Fig. 1c. The Al nanoparticles are found to have a size distribution with an average diameter of 150–250 nm. The inter-planar distance between the fringes is found to be about 0.230 nm (as shown in Fig. 1d) which is consistent with (111) planes of fcc Al nanoparticles.24 Fig. 1e shows the X-ray diffraction patterns of TiO2 and (Au)TiO2 nanohybrid. This indicates that the mixture is composed of mixed anatase and rutile phases, which was in good agreement with the reference patterns of JCPDS card no. 83-2243 and 21-1276, respectively. The tiny peaks at 38.1° (111) (overlapping with the TiO2) and 77.6° (311) in the Fig. 1e are attributed to metallic gold in the (Au)TiO2 nanohybrid. Fig. 1f shows the FESEM image of the (Au)TiO2 nanohybrid, which appeared to be spherical and uniform in size. Since the distribution of Au cannot be clearly shown by the FESEM image, TEM images for (Au)TiO2 nanohybrid were also recorded, and are shown in Fig. 1g. From the Fig. 1g it can be observed that the TiO2 nanoparticles are spherical in shape, with a particle size range of 20–25 nm and the Au nanoparticles size range is 20 nm. The HRTEM images (as shown in Fig. 1h) of (Au)TiO2 nanohybrid show the high crystallinity of the nanoparticles. The inter-planar distance between the fringes is found to be about 0.327 nm and 0.233 nm which is consistent with (110) planes of bulk TiO2 and (111) planes of Au nanoparticles, respectively.25
 |
| | Fig. 1 (a) XRD pattern of Al nanoparticles. (b) FESEM image of Al nanoparticles. (c) TEM image of Al nanoparticles. (d) HRTEM image of Al nanoparticles. (e) XRD pattern of TiO2 and (Au)TiO2 nanohybrid. (f) FESEM image of (Au)TiO2 nanohybrid. (g) TEM image of (Au)TiO2 nanohybrid. (h) HRTEM image of (Au)TiO2 nanohybrid. | |
Fig. 2a shows the UV-visible absorption spectra of TiO2 and (Au)TiO2 nanohybrid. The bare TiO2 nanoparticles show absorption in the UV region at around ∼350 nm (band gap 3.2 eV). Decoration of Au nanoparticles on the surface of the TiO2 significantly influenced the visible light absorption, and an additional peak was also observed at 560 nm due to surface plasmon resonance (SPR) of Au nanoparticles.26 The spectral overlap (J(λ) = 3.91 × 1016 M−1 cm−1 nm4) of the SPR band with the emission spectrum of the dye PP is shown in Fig. 2b revealing the possibility of significant dipolar coupling.27 In Fig. 2c the picosecond resolved emission transients of PP with various nanoparticles are shown. A significant faster component in the case of PP–TiO2 compared to both PP and PP–Al2O3 is clearly evident showing the PET from PP to the host TiO2, which is further enhanced in the presence of Au nanoparticle. The associated time constants are tabulated in Table 1. It has to be noted that the faster fluorescence transient of (Au)PP–TiO2 could be manifestation of FRET from PP to Au nanoparticle in TiO2 matrix. Our control experiment on Au–PP sample shows similar time scales compared to (Au)PP–TiO2. The observation is not confirmatory that the faster fluorescence transient in the latter sample is exclusively for the enhanced electron transfer due to the presence of Au nanoparticles with SPR band rather include the possibility of ground state recovery (recombination) of PP through FRET. However, enhanced photocurrent measurement (see below) concludes the faster fluorescence transient to be due to efficient PET from PP to the host TiO2 in presence of Au Nanoparticles. We have calculated the donor–acceptor distances by following both FRET and NSET mechanisms. Following FRET strategy, we have estimated the donor (PP)–acceptor (surface of Au nanoparticles) distance and it is found to be 8.6 nm, which is within the probing limit of FRET (1–10 nm). Whereas, the donor–acceptor distance following NSET strategy is found to be 3.8 nm which is less than the probing limit of NSET (>10 nm). The calculated FRET distance (8.6 nm) is more close to the radius of the Au nanoparticles (∼10 nm as observed from the HRTEM image) than that of the NSET distance. Thus, FRET over NSET is found to be the prevailing mechanism in this case. The observed PP–Au nanoparticle distance is consistent with the fact that PP would be under the strong influence of Au-SPR for the enhanced electron injection to the TiO2 matrix as evident from other reported literature.27–30
 |
| | Fig. 2 (a) Absorption spectra of TiO2 and (Au)TiO2 nanohybrid. (b) The overlap integral of PP–TiO2 emission and Au absorbance. (c) Fluorescence decay profiles of PP, PP–Al2O3, PP–TiO2, (Au)PP and (Au)PP–TiO2 nanohybrid. | |
Table 1 Lifetimes of picosecond-resolved PL transients of PP, PP–Al2O3, PP–TiO2, (Au)PP, (Au)PP–TiO2, (Al)PP and (Al)PP–TiO2 detected at 630 nm PL maxima upon excitation at 409 nm wavelength. The values in parentheses represent the relative weight percentages of the time components
| System |
τ1 (ps) |
τ2 (ps) |
τ3 (ps) |
τavg (ns) |
| PP |
11400 (100%) |
|
|
11.4 |
| PP–Al2O3 |
10900 (100%) |
|
|
10.9 |
| PP–TiO2 |
300 (59%) |
10000 (41%) |
|
4.2 |
| (Au)PP |
86 (51%) |
9710 (49%) |
|
4.7 |
| (Au)PP–TiO2 |
86 (57%) |
300 (22%) |
7120 (21%) |
1.6 |
| (Al)PP |
11400 (100%) |
|
|
11.4 |
| (Al)PP–TiO2 |
300 (59%) |
9712 (41%) |
|
4.1 |
A comparative study with the Al instead of Au nanoparticles is shown in Fig. 3. The SPR band of the synthesized Al nanoparticles in UV region (∼360 nm) is consistent with reported literature as shown in Fig. 3a.31 A significant fluorescence quenching of PP upon TiO2 and its insignificant change in the proximity of Al nanoparticles are shown in Fig. 3b. The observation demonstrates that Al nanoparticles have insignificant influence in the PET from PP to host TiO2 matrix. At this juncture it has to be noted that enhanced PET in the case of (Au)PP–TiO2 compared to (Al)PP–TiO2 may not be due to the Schottky barrier for the following reason. Following simplified model of White and Catchpole (WC),32 one can estimate the barrier in the case of Al–TiO2 to be lower than that of the Au–TiO2 on taking Al & Au work functions are −4.28 eV & −5.1 eV, respectively and the position of the conduction band of TiO2 is −4.14 eV.33,34 In the above context the rate of PET in the case of (Al)PP–TiO2 is expected to be higher than (Au)PP–TiO2, which is not consistent with our experimental observation (Fig. 2 and 3). However, the light trapping as evident from higher absorption due to the scattering of the Al nanoparticles are evident in Fig. 3c. We have found that 4.6 × 10−7, 5.0 × 10−7, 4.8 × 10−7 and 5.2 × 10−7 mole of dye loading for TiO2, (Al)TiO2, (Au)TiO2 and (Al)(Au)TiO2 electrode with PP whereas 6.0 × 10−7, 6.2 × 10−7, 6.2 × 10−7 and 6.4 × 10−7 mole of dye loading for TiO2, (Al)TiO2, (Au)TiO2 and (Al)(Au)TiO2 electrode with N719.
 |
| | Fig. 3 (a) Absorption spectra of Al nanoparticles. (b) Fluorescence decay profiles of PP, (Al)PP, (PP)TiO2 and (Al)PP–TiO2. (c) Optical absorption of dye-sensitized TiO2 anodes incorporated with/without Al nanoparticles. | |
The photocurrent measurements on the fabricated PP–TiO2 DSSC with Au and Al nanoparticles are shown in Fig. 4. The absorption spectrum of the sensitizing dye is also shown for the comparison. The enhancement of the photocurrent around ∼560 nm in presence of Au compared to Al nanoparticles is evident from the figure. The observation is consistent with the Au-SPR induced electron injection from PP to TiO2 matrix.27,29 The J–V characteristics of the PP–TiO2 DSSC with Au and Al nanoparticles are shown in Fig. 5a. The solar cell parameters are shown in Table 2. From the figure and the table a significant enhancement of efficiency in the Au and Al containing solar cell compared to that of the cell without the metal nanoparticles is evident. The comparable DSSC efficiency in the presence of Al and Au nanoparticles could be rationalized in the following manner. While the SPR in the case of Au nanoparticles in the DSSC helps in the enhanced PET, other factors including light trapping and reduced back electron transfer in the case of Al nanoparticle containing DSSC are responsible for the betterment of efficiency in latter one. As shown in Fig. 5b, the photovoltaic decay in the case of Al nanoparticle containing DSSC is similar to that of the Au nanoparticle containing DSSC, revealing similar less back-electron transfer in the former case.35,36 Thus the scattering of the Al-nanoparticles leading to better light trapping makes the efficiency of the corresponding DSSC comparable to that of the Au-nanoparticle containing DSSC which is consistent with the recent reports.31,37 We have also attempted to fabricate a hybrid PP–TiO2 DSSC containing both Au and Al nanoparticles, where Au and Al nanoparticles are supposed to take care better electron injection and light trapping, respectively. The enhanced solar cell efficiency of the hybrid DSSC is shown in Fig. 5a. Fig. 5b shows slower photovoltage decay in the hybrid solar cell revealing reduced back electron transfer as shown in Table 3. The actual reason for the observation is not clear and needs further investigation which is under way in our laboratory.
 |
| | Fig. 4 (a) Absorption spectra of PP (the inset shows photo of DSSC). (b) Wavelength dependent photocurrent response curves of different DSSCs (base lines were shifted for clarity). | |
 |
| | Fig. 5 (a) Current density–voltage curves under 100 mW cm−2 simulated AM 1.5G solar light irradiation. (b) Open circuit voltage decay profiles of different DSSCs. | |
Table 2 Photovoltaic performance of DSSCs fabricated using different active electrodes sensitized with PP
| Cell |
JSC (μA cm−2) |
VOC (V) |
FF (%) |
Efficiency (%) |
| TiO2 |
1098.09 |
0.490 |
44.56 |
0.22 ± 0.02 |
| (Al)TiO2 |
1345.90 |
0.500 |
51.32 |
0.30 ± 0.01 |
| (Au)TiO2 |
1265.05 |
0.492 |
47.86 |
0.29 ± 0.01 |
| (Au)(Al)TiO2 |
1730.80 |
0.513 |
41.34 |
0.34 ± 0.01 |
Table 3 Dynamics of photovoltage transients of DSSCs fabricated using different active electrodes. The values in parentheses represent the relative weight percentages of the time components
| Active electrode |
τ1 (s) |
τ2 (s) |
τavg (s) |
| PP–TiO2 |
0.16 (78.5%) |
0.50 (21.5%) |
0.23 |
| (Al)PP–TiO2 |
0.11 (75.5%) |
0.80 (24.5%) |
0.28 |
| (Au)PP–TiO2 |
0.12 (68%) |
0.77 (32%) |
0.33 |
| (Au)(Al)PP–TiO2 |
0.12 (66%) |
0.89 (34%) |
0.38 |
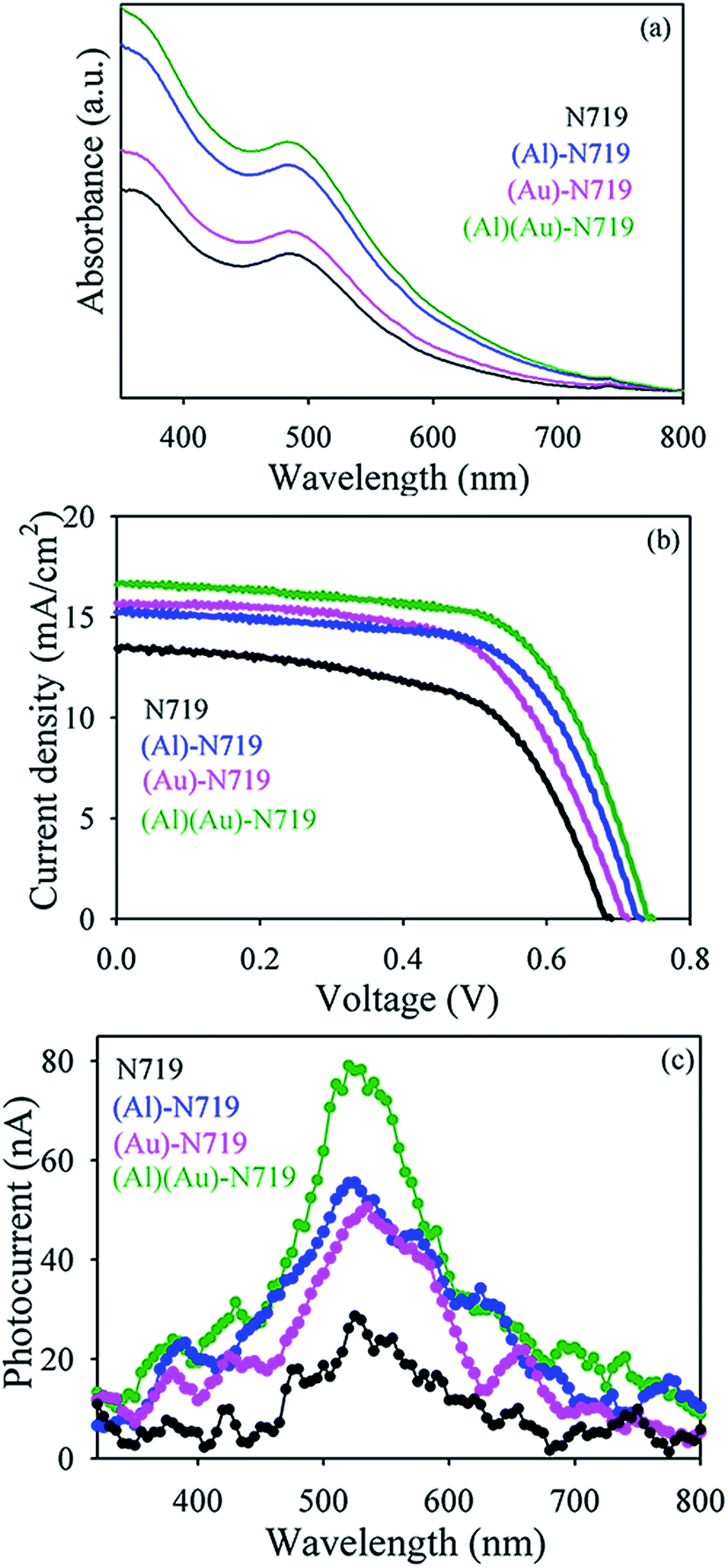
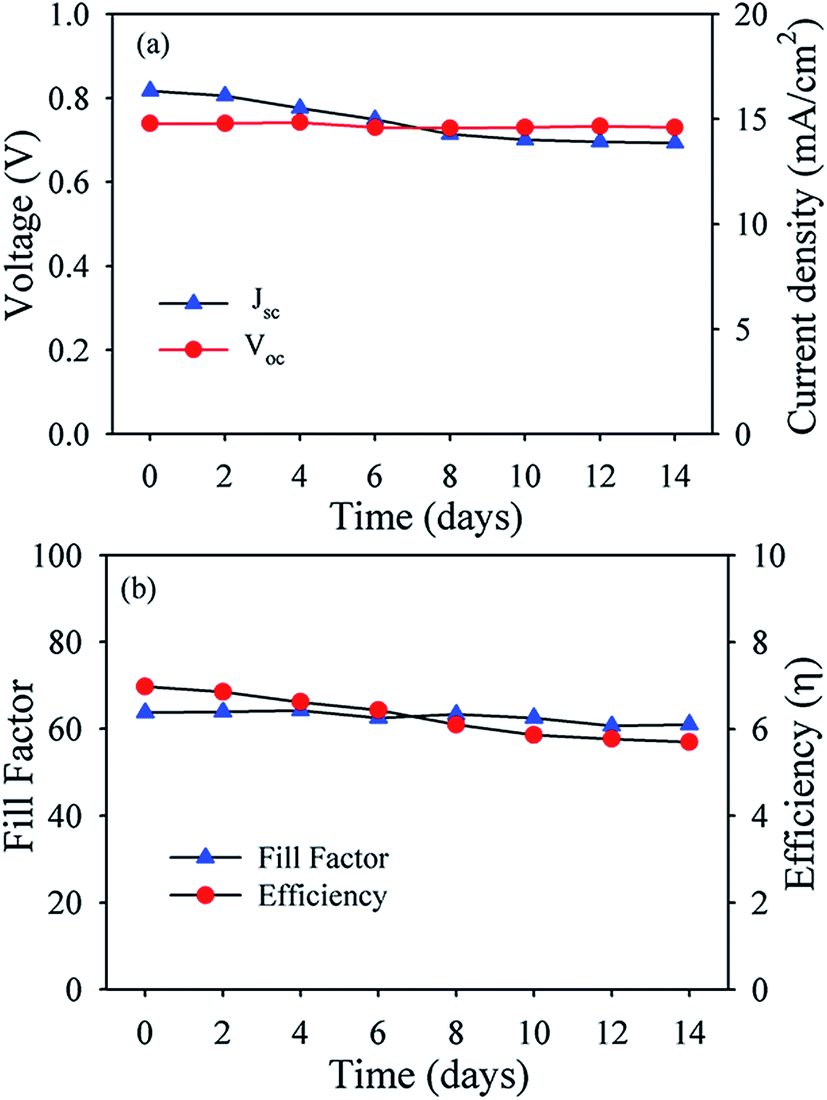
Fig. 6a shows absorption spectra of N719 adsorbed TiO2, (Al)TiO2, (Au)TiO2 and (Al)(Au)TiO2 films. Higher light trapping ability of Al nanoparticles in compare to Au nanoparticles is evident from Fig. 6a. After detail spectroscopic investigation using PP as sensitizer, we developed model prototype device using N719 dye in order to improve efficiencies of DSSCs. The J–V curves obtained for the TiO2 with Au and Al nanoparticles are shown in Fig. 6b. The detailed photovoltaic parameters, including the open-circuit voltage (VOC), short-circuit current density (JSC), fill factor (FF), and PEC are listed in Table 4. The larger PCE of (7.1 ± 0.1)% for DSSC with Au and Al nanoparticles compared to that with TiO2 alone (5.63 ± 0.13)% can be attributed to the fact that plasmonic band and scattering effect are responsible for enhanced electron injection behavior. From Fig. 6c, it is clear that the plasmonic nanoparticles based DSSC exhibits higher photocurrent in the wavelength range of 400–800 nm. Due to dipolar coupling between PP and Au nanoparticles, energy transfer from PP to Au nanoparticles takes place. This leads to transfer of electron from Au nanoparticles to conduction band of TiO2, which increases photocurrent for (Au)TiO2. The light scattering ability by Al nanoparticles enhanced photocurrent for (Al)TiO2. Thus, the improved photocurrent for (Al)(Au)TiO2 may be attributed to the superior light harvesting ability induced by plasmonic and light scattering effect of the Au and Al nanoparticles, respectively. Fig. 7 shows the stability of the dye sensitized solar cells which were kept in an ambient atmosphere. The VOC is very stable during the experimental time window but the efficiency of dye sensitized solar cells began to decrease initially because of the decrease in short circuit current density. This could be due to the instability of the plasmonic nanoparticles in the I−/I3− redox electrolyte or further exposure to the ambient environment.38,39 The overall mechanistic pathways for the enhanced electron injection in DSSC with Au nanoparticles compared to that of Al nanoparticles are schematically shown in Fig. 8. The enhancement in DSSC efficiency in the presence of Au nanoparticles is due to higher light trapping ability by dipolar coupling along with insignificant scattering ability. On the other hand light trapping ability by Al nanoparticles by its significant scattering ability increases the DSSC efficiencies.
 |
| | Fig. 6 (a) Optical absorption spectra of N-719 adsorbed on the TiO2, (Al)TiO2, (Au)TiO2 and (Al)(Au)TiO2 electrode. (b) Current density–voltage curves under 100 mW cm−2 simulated AM 1.5G solar light irradiation. (c) Wavelength dependent photocurrent response curves of different DSSCs. | |
Table 4 Photovoltaic performance of DSSCs fabricated using different active electrodes sensitized with N719
| Cell |
JSC (mA cm−2) |
VOC (V) |
FF (%) |
Efficiency (%) |
| TiO2 |
13.45 |
0.68 |
58.64 |
5.63 ± 0.13 |
| (Al)TiO2 |
15.23 |
0.73 |
63.23 |
6.25 ± 0.1 |
| (Au)TiO2 |
15.68 |
0.71 |
60.47 |
6.05 ± 0.12 |
| (Au)(Al)TiO2 |
16.6 |
0.74 |
63.70 |
7.10 ± 0.1 |
 |
| | Fig. 7 Stability of the device. Plot of (a) JSC and VOC, (b) fill factor and efficiency of the DSSC fabricated using (Au)(Al)TiO2 photoanode with time. | |
 |
| | Fig. 8 Schematic representation of the overall mechanistic pathways for DSSC efficiency in presence of plasmonic nanoparticles (for Au nanoparticles dipolar coupling between Au and PP responsible for higher DSSC efficiency whereas scattering ability for Al nanoparticles responsible for higher DSSC efficiency). | |
4. Conclusion
In summary, we have experimentally demonstrated that plasmonic nanoparticles influence electron injection from a sensitizing dye to the host semiconductor of a model DSSC. While the SPR of Au nanoparticles contribute to enhance the solar cell efficiency through better PET, Al nanoparticles, which have a plasmonic absorption at the UV region essentially, improve solar energy harvesting ability by their better light scattering efficacy. Time-resolved fluorescence technique has been successfully demonstrated that ultrafast electron transfer dynamics are keys for overall electron injection efficiencies. Finally, we have also fabricated and characterized the prototype dye sensitized solar cell in which Au and Al nanoparticles have been employed to engineer the photoanode layer, resulting an increase of power conversion efficiency compared to values achieved in reference devices without Au and Al nanoparticles, respectively. The results highlight the crucial role played by the plasmonic nanoparticles in the plasmonic dipolar coupling as well as scattering effect which are responsible for enhanced solar energy conversation.
Acknowledgements
P. K. thanks Council of Scientific and Industrial Research (CSIR, India) for fellowship. T. K. M. thanks of DST, INSPIRE for fellowship. P. K. S. is thankful to UGC, India for providing the fellowship under the UGC-RGNF scheme. We thank DST, India for financial grant (SB/S1/PC-011/2013). We also thank DAE (India) for financial grant 2013/37P/73/BRNS.
References
- X. Dang, J. Qi, M. T. Klug, P.-Y. Chen, D. S. Yun, N. X. Fang, P. T. Hammond and A. M. Belcher, Nano Lett., 2013, 13, 637–642 CrossRef CAS PubMed.
- W. Hou, P. Pavaskar, Z. Liu, J. Theiss, M. Aykol and S. B. Cronin, Energy Environ. Sci., 2011, 4, 4650–4655 CAS.
- Y. Bai, T. Butburee, H. Yu, Z. Li, R. Amal, G. Q. Lu and L. Wang, J. Colloid Interface Sci., 2015, 449, 246–251 CrossRef CAS PubMed.
- Q. Xu, F. Liu, Y. Liu, K. Cui, X. Feng, W. Zhang and Y. Huang, Sci. Rep., 2013, 3, 2112 Search PubMed.
- J. Yun, S. H. Hwang and J. Jang, ACS Appl. Mater. Interfaces, 2015, 7, 2055–2063 CAS.
- P. Reineck, G. P. Lee, D. Brick, M. Karg, P. Mulvaney and U. Bach, Adv. Mater., 2012, 24, 4750–4755 CrossRef CAS PubMed.
- H. F. Zarick, W. R. Erwin, A. Boulesbaa, O. K. Hurd, J. A. Webb, A. A. Puretzky, D. B. Geohegan and R. Bardhan, ACS Photonics, 2016, 3, 385–394 CrossRef CAS.
- R. Milan, G. S. Selopal, M. Epifani, M. M. Natile, G. Sberveglieri, A. Vomiero and I. Concina, Sci. Rep., 2015, 5, 14523 CrossRef CAS PubMed.
- R. Long and O. V. Prezhdo, J. Am. Chem. Soc., 2014, 136, 4343–4354 CrossRef CAS PubMed.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- S. W. Sheehan, H. Noh, G. W. Brudvig, H. Cao and C. A. Schmuttenmaer, J. Phys. Chem. C, 2013, 117, 927–934 CAS.
- Y. H. Jang, Y. J. Jang, S. T. Kochuveedu, M. Byun, Z. Lin and D. H. Kim, Nanoscale, 2014, 6, 1823–1832 RSC.
- M.-K. Song, P. Rai, K.-J. Ko, S.-H. Jeon, B.-S. Chon, C.-H. Lee and Y.-T. Yu, RSC Adv., 2014, 4, 3529–3535 RSC.
- S. Ramakrishna, M. Pelton, S. K. Gray and T. Seideman, J. Phys. Chem. C, 2015, 119, 22640–22645 CAS.
- P. V. Kamat, J. Phys. Chem. B, 2002, 106, 7729–7744 CrossRef CAS.
- S. D. Standridge, G. C. Schatz and J. T. Hupp, J. Am. Chem. Soc., 2009, 131, 8407–8409 CrossRef CAS PubMed.
- N. Zhou, J. Wang, T. Chen, Z. Yu and G. Li, Anal. Chem., 2006, 78, 5227–5230 CrossRef CAS PubMed.
- Y. Liu and R. Guo, J. Colloid Interface Sci., 2009, 332, 265–269 CrossRef CAS PubMed.
- S. Ghanta and K. Muralidharan, J. Nanopart. Res., 2013, 15, 1715 CrossRef.
- P. Kar, S. Sardar, S. Ghosh, M. R. Parida, B. Liu, O. F. Mohammed, P. Lemmens and S. K. Pal, J. Mater. Chem. C, 2015, 3, 8200–8211 RSC.
- S. Sardar, P. Kar, H. Remita, B. Liu, P. Lemmens, S. K. Pal and S. Ghosh, Sci. Rep., 2015, 5, 17313 CrossRef CAS PubMed.
- S. Sardar, P. Kar, S. Sarkar, P. Lemmens and S. K. Pal, Sol. Energy Mater. Sol. Cells, 2015, 134, 400–406 CrossRef CAS.
- M. A. H. Muhammed, A. K. Shaw, S. K. Pal and T. Pradeep, J. Phys. Chem. C, 2008, 112, 14324–14330 CAS.
- E. Stratakis, M. Barberoglou, C. Fotakis, G. Viau, C. Garcia and G. A. Shafeev, Opt. Express, 2009, 17, 12650–12659 CrossRef CAS PubMed.
- M. Hussain, M. Ahmad, A. Nisar, H. Sun, S. Karim, M. Khan, S. D. Khan, M. Iqbal and S. Z. Hussain, New J. Chem., 2014, 38, 1424–1432 RSC.
- C. Andrei, E. Lestini, S. Crosbie, C. de Frein, T. O'Reilly and D. Zerulla, PLoS One, 2014, 9, e109836 Search PubMed.
- P. Reineck, D. Gómez, S. H. Ng, M. Karg, T. Bell, P. Mulvaney and U. Bach, ACS Nano, 2013, 7, 6636–6648 CrossRef CAS PubMed.
- W. R. Erwin, H. F. Zarick, E. M. Talbert and R. Bardhan, Energy Environ. Sci., 2016, 9, 1577–1601 CAS.
- S. T. Kochuveedu, T. Son, Y. Lee, M. Lee, D. Kim and D. Kim, Sci. Rep., 2014, 4, 4735 Search PubMed.
- K. A. Kang, J. Wang, J. B. Jasinski and S. Achilefu, J. Nanobiotechnol., 2011, 9, 16 CrossRef CAS PubMed.
- X. Chen, B. Jia, Y. Zhang and M. Gu, Light: Sci. Appl., 2013, 2, e92 CrossRef.
- T. P. White and K. R. Catchpole, Appl. Phys. Lett., 2012, 101, 073905 CrossRef.
- H. B. Michaelson, J. Appl. Phys., 1977, 48, 4729–4733 CrossRef CAS.
- Z. Xuming, C. Yu Lim, L. Ru-Shi and T. Din Ping, Rep. Prog. Phys., 2013, 76, 046401 CrossRef PubMed.
- H. Choi, Y.-S. Chen, K. G. Stamplecoskie and P. V. Kamat, J. Phys. Chem. Lett., 2015, 6, 217–223 CrossRef CAS PubMed.
- S. Sardar, S. Ghosh, H. Remita, P. Kar, B. Liu, C. Bhattacharya, P. Lemmens and S. K. Pal, RSC Adv., 2016, 6, 33433–33442 RSC.
- Q. Xu, F. Liu, Y. Liu, W. Meng, K. Cui, X. Feng, W. Zhang and Y. Huang, Opt. Express, 2014, 22, A301–A310 CrossRef PubMed.
- C. Wen, K. Ishikawa, M. Kishima and K. Yamada, Sol. Energy Mater. Sol. Cells, 2000, 61, 339–351 CrossRef CAS.
- J. You, L. Meng, T.-B. Song, T.-F. Guo, Y. Yang, W.-H. Chang, Z. Hong, H. Chen, H. Zhou, Q. Chen, Y. Liu, N. De Marco and Y. Yang, Nat. Nanotechnol., 2016, 11, 75–81 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.