
Enhancing the quality of bio-oil from catalytic pyrolysis of kraft black liquor lignin†
Abstract
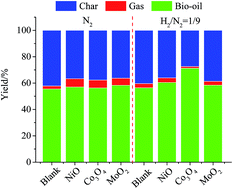
Black liquor is an attractive option for the generation of biofuel and fine chemical intermediates. In this work, kraft lignin was extracted from black liquor and its potential was evaluated in the production of high quality bio-oil via catalytic pyrolysis under two different reaction regimes (N2 and 10% H2 in N2). The 2D HSQC NMR result showed that the lignin obtained directly through acidic precipitation with CO2 mainly consisted of syringyl lignin units and cross-linked C–C bond substructures. The thermal decomposition of the sample mainly occurred at 200–500 °C and the char yield was 44.46% at 700 °C. The catalytic pyrolysis experiment indicated that the addition of catalysts (NiO, MoO2, and Co3O4) could inhibit char formation and enhance the properties of bio-oil by removing oxygenated compounds via releasing CO and CO2 under a N2 atmosphere. The addition of the catalysts could prompt lignin degradation and increase the yield of bio-oil in the presence of H2. The yield of bio-oil increased by 26.38% with Co3O4 catalytic pyrolysis under a H2/N2 atmosphere.
 Please wait while we load your content...
Please wait while we load your content...

